Doğumsal kusurlar ve anomaliler. Doğum kusurları: nedenleri, önlenmesi ve tedavisi
Aşağıdaki doğum kusurlarından bazıları oldukça iyi bilinirken, diğerleri son derece nadirdir. Ama ne olursa olsun, hepsi oldukça ürkütücü, gizemli ve trajik. Yani…
Bu, ikizlerin vücudun bir veya daha fazla bölümünde birleşik olarak doğduğu nadir bir durumdur (yaklaşık 200.000 doğumda bir). Tüm vakaların %70-75'inde Siyam ikizleri dişidir. Yaklaşık yarısı ölü doğar. Bazen ayrılabilirler, bu da izin verir Siyam ikizleri canlı tüm hayat ancak çoğu zaman bu mümkün değildir.
Hipertrikoz (Ambrams sendromu)
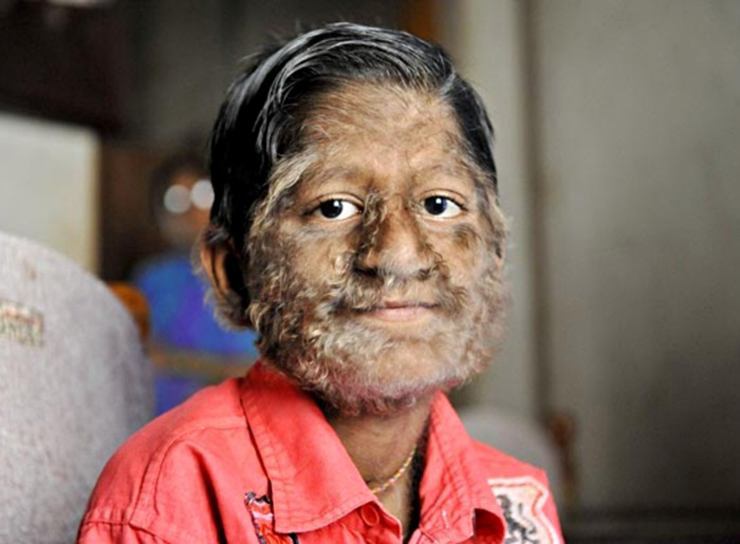
Hipertrikoz, karakterize bir hastalıktır. aşırı büyüme saç, cildin bu bölgesi için alışılmadık. Neyse ki, bu çok ve şu anda dünyada hipertrikozdan muzdarip sadece 40 kişi var. Akranları tarafından genellikle reddedildikleri için hastalık çocuklar için son derece zayıflatıcıdır.
Sirenomelia (deniz kızı sendromu)
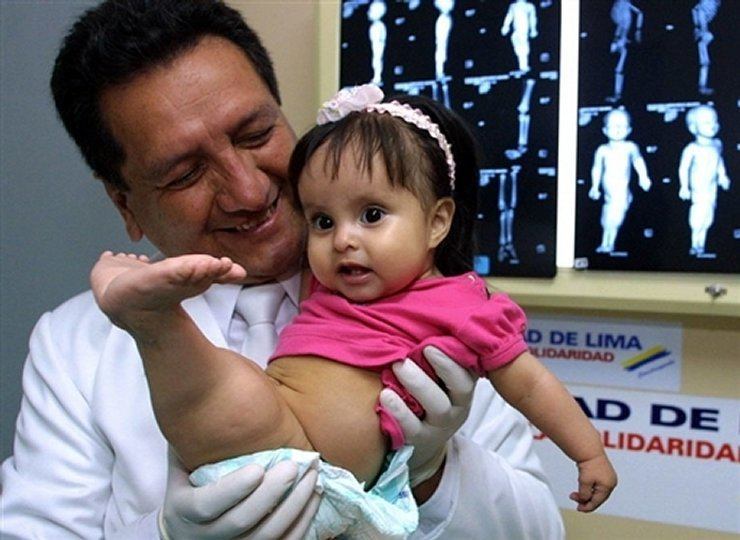
Sirenomelia, füzyon şeklinde kendini gösteren bir gelişimsel anomalidir. alt ekstremiteler. 100 bin yenidoğanda bir vakada görülür. Kural olarak, doğumdan 1-2 gün sonra ölüme yol açar, bu böbreklerin gelişimi ve işleyişindeki tuhaflıklardan kaynaklanır ve Mesane. Bununla birlikte, bu anomaliye sahip çocukların (hatta cerrahi müdahale) birkaç yıl yaşadı. Böylece sirenomelia'dan muzdarip Amerikalı kız Shiloh Pepin, 10 yıldan fazla yaşayabildi.
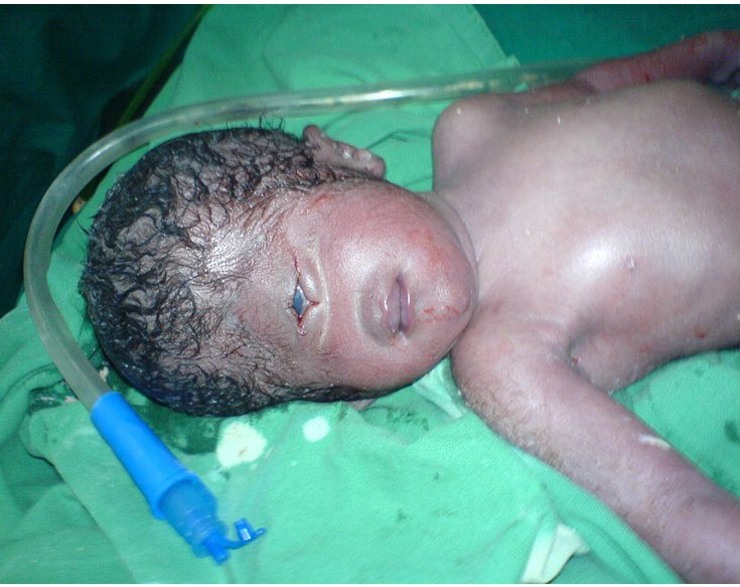
Cyclopia, tahmin edebileceğiniz gibi, adını ünlü efsanevi yaratık Cyclops'tan almıştır. Siklopi ile doğan çocukların, başın ortasında yer alan tek gözü vardır. Tüm vakaların %100'ünde yenidoğanlar yaşamın ilk günlerinde ölür.
Kafaya hangi ikizlerin füzyon tipi normal çocuk gövdesi olmayan ikizin başı uzar. Tarih, bu anomalinin yalnızca on kayıtlı örneğini biliyor ve bunlardan yalnızca üçünde çocuk doğumdan sonra hayatta kaldı. Bir vakada, ikinci kafa gülümseyebiliyor, gözlerini kırpabiliyor, ağlayabiliyor ve annenin memesini emebiliyordu.
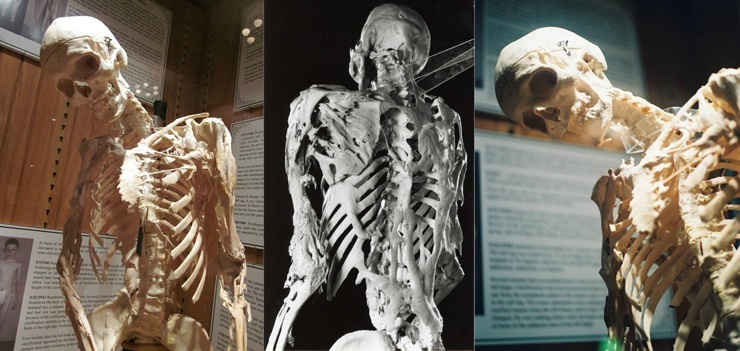
En nadir hastalık(2 milyonda 1 vaka), bir gen mutasyonundan kaynaklanır ve doğuştan gelişimsel kusurlarla kendini gösterir - öncelikle kavisli başparmak durdurma ve ihlaller servikal bölge omurga. Fibrodisplazinin temeli oluşumudur. inflamatuar süreçler tendonlarda, bağlarda, fasyada, aponevrozlarda ve kaslarda sonuç kalsifikasyon ve ossifikasyona yol açar. Hastalığa "ikinci iskeletin hastalığı" da denir, çünkü aslında vücutta düzenli anti-inflamatuar süreçlerin meydana gelmesi gereken yerlerde kemik büyümesi başlar.
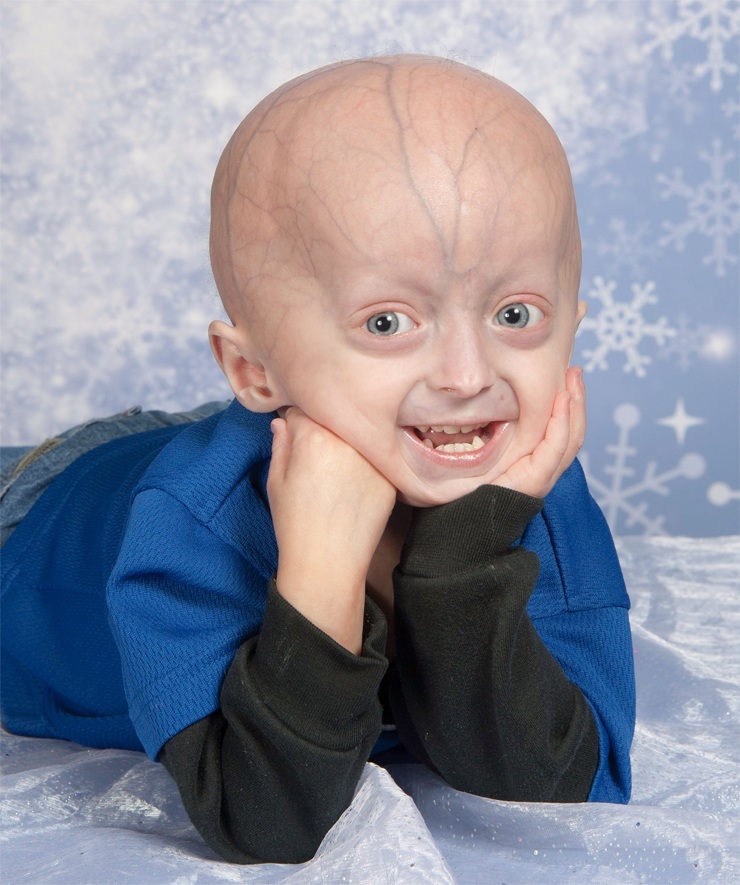
Progeria - en nadir genetik kusur cilt değişikliklerinin meydana geldiği ve iç organlar, Nedeniyle erken yaşlanma organizma. Dünyada 80'den fazla progeria vakası kaydedilmemiştir.
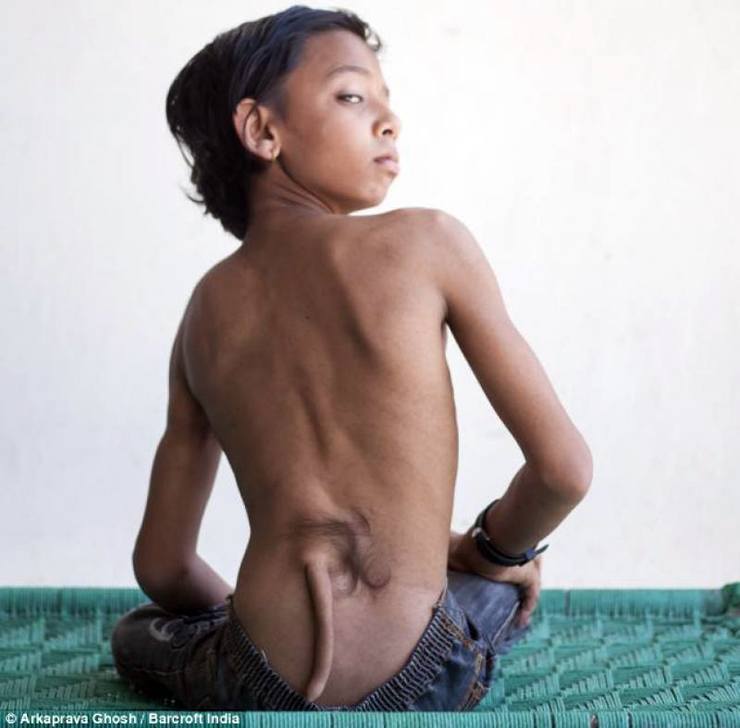
doğum kusuru Bebeğin kasları, sinirleri, derisi ve diğer organları ile birlikte yarı işlevsel bir kuyrukla doğduğu yer. kan damarları. Bir gen mutasyonundan kaynaklandığı düşünülmektedir.

Anensefali - tam veya kısmi devamsızlık yarım küreler beyin, kafatası kemikleri ve yumuşak dokular. Yaklaşık 10 bin yenidoğanda bir (ABD'de), daha sık olarak kız fetüslerinde görülür. Vakaların %100'ündeki kusur ölümcüldür. Anensefali olan fetüslerin %50'si rahimde ölür, geri kalan %50'si canlı doğar, ancak yalnızca %66'sı birkaç saat yaşayabilir (ancak bazılarının yaklaşık bir hafta yaşadığı durumlar vardır). Daha çok Bebek Kay lakabıyla tanınan Stephanie Keane, 2 yıl 174 gün bu korkunç teşhisle yaşayan anensefaliler arasında "uzun ömürlü" olarak kabul edilir.
Sosyal paylaşım ağlar
Yenidoğanların yaklaşık %2-3'ünde ciddi konjenital malformasyonlar vardır. Embriyolojik olarak, bu tür kusurlar üç ana sınıfa ayrılır (Tablo 36-6):
doğum kusurları eksik morfogenezin bir sonucu olarak;
tekrarlayan morfogenezin bir sonucu olarak doğum kusurları;
Anormal morfogenezin bir sonucu olarak doğum kusurları. Eksik morfogenez en yaygın patolojidir, anormal - en nadirdir.
Tablo 36-6.
(Cohen MM, 1997)
Hemen hemen tüm doğumsal malformasyonlar embriyonik dönem(gebeliğin 3-10. Haftası), organların farklılaşmasının olduğu dönemde (Tablo 36-7).
Doğum kusurları basit veya karmaşık olabilir. Nasıl daha sonraki son tarih doğuştan bir kusurun ortaya çıkması, büyük olasılıkla bitişik embriyonik yapılarda (basit konjenital malformasyonlar) patogenetik olarak ilgili kusurların olmayacağı. Embriyogenezin erken evrelerinde bir kusur oluşursa, bitişik yapıların etkilenme olasılığı oldukça yüksektir ve birden fazla doğum kusurları konjenital malformasyonların gelişimi veya sırası (sırası). Bir örnek, intrauterin hipoplazi şeklinde birincil kusur olduğunda Pierre Robin dizisidir. çene kemiği dilin aşağı indirilmesi sürecinin ihlaline neden olur ve bu da damağın kapanmamasına yol açar.
Tablo 36-7. Konjenital malformasyonların oluşum zamanı (Cohen M.M., 1997)
konjenital malformasyon onun klinik tezahür minimal (küçük dilin çatallanması) ve maksimum derecede belirgin (damağın tıkanmaması) olabilir. Minimal tezahür durumunda, şu şekilde tanımlanacaktır: küçük anomali gelişim. Kompleks bir malformasyon veya sekans da minimal olarak mevcut olabilir. klinik değişken.
Bu nedenle, örneğin, en şiddetli versiyonundaki holoprosensefali dizisi, serebral hemisferlerin konjenital malformasyonu ve yüz anomalileri ile karakterize edilir - burun yapılarının yokluğu, hipotelorizm, yarık dudaklı premaksiller agenezi ve Alveolar süreç üst çene; minimal bir klinik varyantta, tek bir maksiller kesici ile hipotelorizm kombinasyonu ile karakterizedir. Bunu bilmek, holoprosensefali olan bir çocuğun aile genetik prognozu için son derece önemlidir - ebeveynleri minimal klinik belirtiler açısından incelemek.
DEFORMASYONLAR
Bu tip doğum kusuru yenidoğanların yaklaşık %1-2'sinde bulunur. En sık görülen kusurlar çarpık ayak, doğuştan kalça çıkığı ve pozisyonel skolyozdur (postural skolyoz). Deformiteler en sık olarak geç fetal dönemde üç ana neden ve predispozan faktörün bir sonucu olarak ortaya çıkar (Tablo 36-8): mekanik nedenler; konjenital malformasyonlar; işlevsel nedenler.
Deformitelerin mekanik nedenleri en yaygın olanıdır ve fetal hipokinezinin arka planında ortaya çıkar. 4.500 yenidoğan üzerinde yapılan bir çalışmada, tüm yenidoğanlarda deformitesi olan çocukların 1/3'ünde iki veya daha fazla deformite olduğu gösterilmiştir. Bu dizi doğuştan şekil bozuklukları Uterus sertliğinin üç deformiteye neden olduğu bir örnekle iyi bir şekilde gösterilmiştir: plajiyosefali, alt çene asimetrisi ve bir yenidoğanda çarpık ayak.
Tablo 36-8. Deformitelerin gelişiminde predispozan faktörler (Cohen M.M., 1997)
| Mekanik | |
| nedenler | Rahim ve karın kaslarının sertliği (ilişkili |
| ilk doğum) | |
| Hamile bir kadının kısa boyu ve küçültülmüş vücut boyutu | |
| kadın | |
| hipoplazi pelvik halka | |
| Rahim hipoplazisi | |
| iki boynuzlu rahim | |
| Rahim leyomiyomu | |
| Olağandışı implantasyon bölgesi | |
| Kronik amniyotik sıvı kaçağı | |
| oligohidramnios ( çeşitli etiyolojiler) | |
| Olağandışı fetal pozisyon | |
| Fetal başın erken pelvik yerleştirilmesi | |
| Çoklu hamilelik | |
| Fetusun konjenital malformasyonları | |
| büyük meyve(doğuştan makrozomi) | |
| Makrosefali veya fetal hidrosefali | |
| doğum kusurları | Spina bifida; |
| olarak fetal gelişim | |
| deformasyonların nedeni | Diğer konjenital malformasyonlar gergin sistem cenin |
| Fetal böbrek agenezisi (bilateral) | |
| Şiddetli böbrek hipoplazisi | |
| Şiddetli polikistik böbrek hastalığı | |
| üretral atrezi | |
| fonksiyonel | nörolojik bozukluklar(doğuştan hipotansiyon) |
| nedenler | |
| Kas bozuklukları | |
| Kusurlar bağ dokusu | |
Hemen hemen tüm ciddi doğum kusurları idrar sistemi sırayla Potter sendromunun (fetüsün olağandışı yüzü, uzuvların çoklu kontraktürleri) nedeni olan oligohidramnios'a neden olur.
Deformitelerin fonksiyonel nedenleri şunları içerir: çeşitli formlar yenidoğanların konjenital hipotansiyonu ve nöromüsküler artrogripoz türleri. Konjenital hipotansiyon, mikrognati, mikroglossi, çıkıntılı lateral sütürler ile ilişkili olabilir. Sert damak el ve ayağın anormal fleksiyon kıvrımları, düz valgus ayaklar ve diğer şekil bozuklukları. Artrogripozis, uzuvların konjenital sertliği ve eklemlerin karakteristik bir pozisyonda sabitlenmesi ile karakterizedir.
KESİNTİ
Bozulmaların kesin sıklığı bilinmemektedir, yenidoğanların %1-2'sinde saptanır. açıklayan ilk araştırmacıdır. bu tür 1968 tarihli "Gebelik Sırasında Amniyon Rüptürünün Neden Olduğu Fetal Malformasyonlar" monografisindeki patoloji, R. Torpin'di (Cohen M.M., 1997). Maruz kalma sonucu bozulmalar ortaya çıkar çeşitli sebepler: vasküler faktörler, anoksi, enfeksiyonlar, radyasyon, teratojenler, amniyotik bantlar, mekanik faktörler. Bozulmaların tipi ve ciddiyeti gebelik yaşına, darbenin yerine ve doku hasarının derecesine bağlıdır. Çoğu zaman, fetal dönemde bozulmalar meydana gelir, ancak teratojenik etkiler, embriyonik morfogenezin karakteristiğidir. Bu maruziyetlerden bazıları, embriyonik dönemde ortaya çıkan “fenokopi” konjenital malformasyonlardır. Örneğin, amniyotik bantlar erken dönem gebelikler anensefali, yarık dudak ve damak, ekstremitelerde redüksiyon kusurlarına neden olabilir. en zor ayırıcı tanı konjenital malformasyon ile damar patolojisi sonucu bozulma arasında (Tablo 36-9).
Tablo 36-9. Embriyo ve fetüste vasküler bozulma mekanizmaları (Cohen M.M., 1997)
| patogenez | yapısal anomali |
| Embriyonun yok edilmesi kılcal ağ | Erken amniyotik sıralama, uzuv-gövde duvarı kompleksi, uzuv redüksiyon anomalileri, maksiller ve uzuv hipoplazisi |
| Embriyonik damarların kalıcılığı | Uzuv kusurları: radyal aplazi, tibial aplazi, fibular aplazi, çarpık ayak |
| Embriyonik damarların erken amputasyonu | Patoloji dizisi Subklavyan arter(Polonya, Möbius, Klippel-Feil sekansları), gastroşizis, at nalı böbrek |
| Vasküler olgunlaşmanın ihlali | Kapiller hemanjiyomlar, arteriyovenöz fistüller, anevrizmalar (Berry anevrizmaları) |
| Kan damarlarının tıkanması (dış kompresyon) | Leiomyomlarla ilişkili anomaliler, tubal gebelik ve bikornuat uterus |
| Kan damarlarının tıkanması (embolik tromboz) | Porensefali, hidranensefali, mikrosefali, safra kesesi atrezisi, distal sindaktili, hemifasiyal mikrozomi (nadir), bilateral anorşim, deri aplazisi |
| Hemodinamik ihlali | Hamilelik sırasında kokain kullanımının neden olduğu anomaliler |
İzole doğuştan gelişimsel kusurlar teşhiste zorluk çıkarmaz. İzole doğum kusurları olan çocukların teşhis ve tedavisine ilişkin ampirik deneyim ve bilginin yalnızca yetersiz değil, aynı zamanda sıklıkla hatalı olduğu çoklu doğum kusurları alanında tamamen farklı bir durum gözlemlenmektedir.
Klinik uygulamanın ihtiyaçları, çoklu konjenital gelişimsel kusurların etiyolojisini ve patogenezini incelemek için araştırma çalışmalarının genişlemesine katkıda bulunmuştur. Bu bölüm daha sonra sendromoloji olarak adlandırıldı. Sendromoloji ve klasik tıp arasındaki farkın açık örneklerinden biri, yani. Bir organ veya sistemin izole bir patolojisinin teşhis edildiği ve incelendiği tıp, klasik tıp 20. yüzyılda sadece birkaç yeni hastalık tanımlanmıştır ( radyasyon hastalığı, Lejyoner hastalığı, AIDS, Lyme hastalığı), sendromolojide bu tür nozolojik formların sayısı 5000'i aştı ve her yıl en az 80 yenisi tanımlanıyor.
Bazı sendromik patoloji biçimleri için, modern moleküler genetik, onları belirleyen genlerin lokalize edilmesini ve daha çok zar reseptörleri veya doku büyüme faktörleri tarafından temsil edilen gen transkripsiyon ürünlerini incelemeyi mümkün kılmıştır. Örneğin, Hirschsprung hastalığında, farklı genlerin iki mutasyonu tanımlanmıştır: RET onkogen ve bu konjenital patolojinin iki genetik tipini ayırt etmeyi mümkün kılan endotelin B reseptörü.
Sendromoloji, tıbbın neredeyse tüm uzmanlık alanlarını kapsayan son derece geniş bir alandır. Tüm yeni doğan bebeklerin yaklaşık %1'inde birden fazla Doğuştan anomaliler veya sendromlar. Bunlardan, vakaların %40'ında, belirli bir sendrom bugün teşhis edilebilmektedir ve geri kalan %60'ının “yeni” sendromlar olarak tahsis edilmesi gerekmektedir. Pek çok sendrom oldukça nadir olmakla birlikte, toplam olarak sendromik patoloji formları tıbbın niceliksel olarak önemli bir bölümünü oluşturur.
Tablo 36-10. Embriyo ve fetüste vasküler bozulma mekanizmaları (Cohen M.M., 1997)
| Çevresel faktörler | teratojenik etki |
| Farmakolojik müstahzarlar | |
| Talidomid | Ekstremitelerin redüksiyon anomalileri. Üst ekstremite kuşağının hipoplazisi. |
| Kulak anomalileri | |
| Alkol | Gecikme fiziksel Geliştirme. Alışılmadık fenotip (kısa göz kapakları) |
| yuvalar). Mikrosefali, mental retardasyon | |
| DietylstyleOestrol | Vajinal adenomatozis. Servikal erozyon. vajinal adenokarsinom |
| (seyrek) | |
| varfarin | |
| Burun kıkırdaklarının hipoplazisi. Merkezi sinir sisteminin doğuştan kusurları. Nokta kalsifikasyonu | |
| epifizler | |
| Hidantoin (dilantin) | |
| Gecikmiş fiziksel gelişim. Olağandışı fenotip. Mikrosefali, zihinsel | |
| geri kalmışlık | |
| trimethadion | |
| Gecikmiş psikomotor gelişim. Olağandışı fenotip (kemerli | |
| kaşlar). Dudak veya damak enfeksiyonu | |
| aminopterin | |
| metotreksat | Spontan kürtajlar. konjenital hidrosefali. Gecikmiş fiziksel gelişim |
| tiya. Olağandışı fenotip | |
| Streptomisin | |
| Konjenital sensörinöral işitme kaybı | |
| tetrasiklin | |
| konjenital hipoplazi diş minesi Dişlerde renk değişikliği (sarı dişler) | |
| sodyum valproat | |
| Nöral tüp defektleri (Spina bifida) | |
| retinol | |
| Spontan kürtajlar. Kraniyofasiyal anomaliler. Nöral tüp defektleri | |
| lityum müstahzarları | |
| Doğuştan kalp kusurları (Ebstein anomalisi) | |
| Antitiroid ilaçlar | |
| VG. Guatr | |
| Androjenler ve yüksek dozlar | |
| erkekleştirme | erkekleştirme |
| progestinler | |
| penisilamin | |
| Hiperelastik cilt. doğuştan patoloji bağ dokusu | |
| Metilcıva (cıva) | Kimyasal maddeler |
| doğuştan atrofi beyin. Spastisite, konvülsiyonlar. zihinsel | |
| geri kalmışlık | |
| Öncülük etmek | |
| Gecikmiş fiziksel gelişim. Olağandışı cilt rengi (gri) | |
| Sigara içmek | Fiziksel, beslenme ve diğer etkiler |
| Spontan kürtajlar. IUGR | |
| iyonlaştırıcı radyasyon | |
| Hasar gebelik yaşına bağlıdır. Spontan kürtajlar. Doğuştan | |
| gelişimsel kusurlar (gebeliğin 18-36. Günü). Mikrosefali ve mental retardasyon | |
| Kayıp (gebeliğin 8-15. Haftası) | |
| Yüksek ateş | |
| CNS'nin doğum kusurları | |
| anne diyabeti | |
| UPU. Kaudal Regresyon Sendromu | |
| Gıdalarda iyot eksikliği | |
| annede PKU | Guatr. Zeka geriliği ve gecikmiş fiziksel gelişim |
| Kürtaj, mikrosefali, mental retardasyon |
Semptomlar veya semptom kompleksi tek bir nedene bağlıysa (tek nedenli etiyoloji), o zaman sendrom terimi, hastalığın nozolojik biçimini (nozolojik sendrom) ifade eder ve bu anlamda "hastalık" terimi ile eşanlamlıdır. Uygulamada, uluslararası uzmanların tavsiyelerini takiben, hastalığın ilerleyici seyri olan durumlarda "hastalık" terimi daha iyi kullanılır.
Bu nedenle, sendrom etiyolojik olarak belli bir hastalık pleiotropik (çoklu) bir etki ile.
Böyle bir sendromun bir örneği, Recklinghausen hastalığı veya tip 1 nörofibromatozisin izolasyon öyküsüdür. 1849'da, Dublin Tıp Okulu'nun önde gelen cerrahlarından Robert Smith, iki nörofibromatozis vakasının klinik ve patoanatomik özelliklerini yayınladı ve daha önceki 75 yayından verileri aktardı. tıp literatürü. Bununla birlikte, yalnızca Recklinghausen'in (von Recklinghausen, 1882) mesajında, nozolojik bir form olarak nörofibromatoz fikri doğrulandı. Artık bu patolojinin en yaygın insan kalıtsal hastalıklarından biri olduğu ve 2000 doğumda 1 sıklıkta ortaya çıktığı gösterilmiştir. Modern teşhis kriterleri bu hastalık temelinde karakteristik semptomlar derinin hiperpigmentasyonu ("café au lait" gibi), doğuştan yanlış eklemler veya alt ekstremite kemiklerinin eğriliği gibi, ancak 1987'de geliştirilmiştir. Aşağıdaki belirtilerden ikisi ve bunların başka bir hastalığın belirtisi olmaması şartıyla.
Nörofibromatozis tip 1 (Recklinghausen hastalığı) için tanı kriterleri (WHO bildirisi, 1992):
Ergenliğe girmemiş bir hastanın suni ışık altında yapılan muayenesinde en az beş adet açık kahverengi pigment lekesi (en geniş yeri 5 mm'den fazla) bulunur; ergenliğe ulaşmış bir hastayı muayene ederken - en az 6 yaşlılık noktası (en geniş noktada 15 mm'den fazla);
Anamneze göre varlığı veya Klinik muayene herhangi bir tipte iki veya daha fazla nörofibrom veya bir pleksiform nörofibrom;
Çoklu, çil benzeri karanlık noktalar koltuk altı veya kasıkta;
kanat displazisi sfenoid kemik ya da doğuştan eğrilik ya da uzun kemiklerin incelmesi ile şekillenmesi yanlış eklem veya onsuz;
Optik sinirin gliomu;
Yarık lamba incelemesinde iris üzerinde saptanan iki veya daha fazla Pisch lekesi/nodülü;
Birinci derece akrabada (ebeveyn, kardeş veya çocuk) yukarıdaki kriterlere göre nörofibromatozis tip 1 varlığı.
Recklinghausen nörofibromatozunun zamanında teşhisi şunları gerektirir: dinamik gözlem başın periyodik BT taraması ile ve omurilik amacıyla erken teşhis CNS neoplazisi.
Zaten XX yüzyılın 60-70 yıllarında. çoğu kromozomal ve teratojenik sendrom tanımlanmış ve büyük miktar gen sendromları. XX yüzyılın 80'lerinin başında. deneyim, yalnızca birleşik bir uluslararası terminoloji geliştirmeye değil, aynı zamanda "yeni" sendromları tanımlamak ve çoklu gelişimsel kusurların patogenezini incelemek için bazı metodolojik yaklaşımlar önermeye izin verdi (Spranger J. ve diğerleri, 1982; Cohen MM., 1982; Cohen MM. , 1997).
Uygulamada, sendromik patoloji formları, tek bir konjenital kusura ek olarak, bir çocukta olağandışı bir fenotipin, yani üç veya daha fazla küçük gelişimsel anomalinin varlığının kaydedildiği durumları da içerir.
Küçük gelişimsel anomaliler veya disembriyogenez damgaları anormal morfolojik varyantlardır. bireysel organlar veya sahip olmayan kumaşlar tıbbi değer, yani tedavi gerektirmeyen. Bu varyantların oluşumu, embriyonik veya daha nadiren doğurgan dönem insan morfogenezi. AT klinik genetik ve sendromoloji, küçük gelişimsel anomaliler son derece önemli bir teşhis işaretidir ve yüksek bir olasılığa işaret eder. ciddi ihlaller gerektiren konjenital malformasyonlar şeklinde morfogenez özel teşhis ve sıklıkla sonraki cerrahi müdahaleler (Tablo 36-11).
İnsanlarda 200'den fazla bilgilendirici morfogenetik varyant tanımlanmıştır. klinik uygulama genellikle 80'den fazla küçük gelişimsel anomali kullanılmaz.
Yenidoğanlarda küçük gelişimsel anomaliler:
Kafa:
V anormal saç büyüme modeli;
V düzleştirilmiş oksiput;
Kafatası kasasının V "yumruları".
Yörünge alanı:
V epikantik kıvrımlar;
V ters epikant;
Gözlerin V Mongoloid bölümü;
Gözlerin V anti-Mongoloid bölümü;
V kısa palpebral fissürler;
Gözün dış köşelerinin V distopisi;
V orta derecede hipotelorizm;
V orta derecede hipertelorizm;
Pitoz hafiftir;
irislerin V heterokromisi;
V mikrokornea.
Kulak kabukları:
V ilkel form;
V Darwin tüberkülü;
V kıvrımın anormal şekli;
V asimetrik kulak kepçeleri;
V döndürülmüş kulak kepçeleri;
V azaltılmış kulak kepçeleri;
V çıkıntılı kulak kepçeleri;
V tragus olmaması;
idrar bölünmesi;
V lob yokluğu;
V kulak çukurları;
Kulak "çıkıntıları";
Dış işitsel kanalın V daralması.
Burun ve filtre:
¦¦¦ burun kanatlarının hipoplazisi;
¦¦¦ konuşlandırılmış burun delikleri;
¦¦¦ düzleştirilmiş filtre;
¦¦¦ çıkıntılı filtre.
ağız bölgesi ve ağız boşluğu:
¦¦¦ mikrogeni;
¦¦¦ dilin yarılması;
¦¦¦ ağız deliğinin anormal frenulumu;
¦¦¦ yenidoğan diş filtresi.
¦¦¦ pterygoid boyun - orta derecede;
¦¦¦ boyun fistülleri.
İlkel polidaktili; avuç içi tek fleksiyon katı; anormal dermatoglifikler; küçük parmakların klinodaktili; 4-5 parmakların kısalması; hipoplastik terminal falankslar.
eşzamanlı olarak parmaklar; sandalet boşlukları; kısa 1. parmak; parmakların yerleştirilmesi (IV-V); kalınlaşmış tırnaklar
Cilt kapakları:
hemanjiyomlar;
Cilt hiperpigmentasyonu ve benleri; Moğol lekeleri (beyaz ırkta); cilt depigmentasyonu; aksesuar meme uçları veya areolalar.
Gövde:
¦¦¦ rektus abdominis kaslarının diyastazı;
¦¦¦ orta derecede hipospadias (başlar);
¦¦¦ sakrumun derin izlenimleri.
iskelet:
¦¦¦ sternumun çökmesi veya çıkıntısı.
Baş, boyun ve el bu belirtilerle ilgili olarak en bilgilendiricidir, tüm küçük gelişimsel anomalilerin% 70'inden fazlası bu bölgede bulunur. Teşhis değeri küçük gelişimsel anomaliler farklıdır. Bu işaretlerin pratik öneminin üç veya daha fazla anomalinin teşhisinde yattığı temel olarak önemlidir. Üç veya daha fazla küçük gelişimsel anomalisi olan her yenidoğanın beyin, kalp, böbrekler veya omurgada ciddi doğumsal malformasyon olasılığı yüksektir (%90) yüksek olasılık(% 50) patolojinin sendromik formunun teşhisi. Psikomotor gelişim hızında gerilik olan bir çocuğun üç veya daha fazla küçük gelişimsel anomalisi varsa, yüksek risk organik hasar CNS (bkz. Tablo 36-11). Bazen sadece iki küçük gelişimsel anomalinin varlığı tanı için bilgilendiricidir. Örneğin, hipotelorizm (yakın aralıklı gözler) ve tek üst kesici prosensefalik grup gibi beynin konjenital bir malformasyonunu gösterir.
Bir çocukta doğuştan gelişimsel bir kusurun teşhisi, neonatoloğa aşağıdaki soruları sorar:
Bu kusur ne tür bir patolojiye aittir (doğuştan malformasyon, bozulma, şekil bozukluğu veya displazi)?
Bu doğum kusuru ile diğer doğum kusurları veya henüz klinik olarak tezahür etmeyen hastalıklar şeklinde ilişkiler ne sıklıkla vardır?
Bu konjenital kusur hangi sıklıkla patolojinin sendromik formunun bir belirtisidir?
Bu konjenital defektte en sık görülen sendromlar nelerdir?
Bu soruların cevapları ilk sırada teşhis aşaması pratik ortak çalışma neonatolog ve genetikçi. Nihai hedef bu aşama, ek doğuştan gelişimsel kusurların teşhisi veya belirli bir sendromun teşhisidir. Sendromik bir patoloji formunun teşhisi durumunda, çoğu durumda daha da netleşir. tıbbi taktikler muhafazakar veya cerrahi tedavi ve hasta bir çocuğun ailesinde tıbbi genetik prognoz. Belirli bir sendromda yaşam ve sağlık için prognoz hakkında bilgi büyük önem ve tıbbi çalışmanın temel amacıdır.
Sendromlar, çeşitli biyolojik organizasyon düzeylerinde analiz edilebilir:
Metabolik süreçlerdeki bozukluklar düzeyinde (dismetabolik sendromlar);
Doku bozuklukları düzeyinde (displazi sendromları);
Organ morfolojisi bozuklukları düzeyinde (konjenital malformasyon sendromları ve bozulma sendromları);
Vücudun belirli bir bölümünün ihlalleri düzeyinde (deformasyon sendromları). Dört ihlal seviyesinin hepsinde ve pratik değer, çünkü dört biyolojik sendrom tipinin tümü, sağlık için tanı ve prognoz için net tanısal ve genetik kriterlere sahiptir (Tablo 36-12).
Yenidoğanda sendromik patoloji formlarının teşhisi şunları içerir:
Olasılık doğru teşhis eşlik eden hastalıklar birden fazla kusuru olan bir çocuk;
Cerrahi veya cerrahi sırasında her sendromun özelliği olan komplikasyonların prognozu konservatif tedavi doğum kusurları veya hastalıkları;
Hastalıkların cerrahi veya konservatif tedavi olanaklarının doğru bir şekilde değerlendirilmesi (cerrahi müdahalenin zamanlaması ve kapsamı, uzun vadeli sonuçlar tedavi);
Ailede doğru tıbbi genetik prognoz.
Bu bulgular, klinik uygulamadan elde edilen aşağıdaki gözlemlerle iyi bir şekilde gösterilmektedir.
Tablo 36-12. biyolojik tipler sendromlar (Cohen MM, 1982)
| Sendrom tipi (bozukluk düzeyi) | Karakteristik özellikler | örnekler |
| Dismetabolik sendrom (metabolizma) Displazi sendromu (doku) | Yenidoğanlar progradyan bir klinik ile normal bir fenotipe sahiptir. Klinik işaretler diğer sendrom tiplerine kıyasla nispeten benzerdir. Konjenital malformasyonlarla ilişkisi yok. Birincil biyokimyasal kusurun doğrulanması mümkündür. Otozomal resesif kalıtım Basit displazi sendromu sadece lezyonlarla karakterizedir | PKU, Tay-Sachs hastalığı, HCV Marfan sendromu, Ehlers-Danlos sendromu, akondroplazi |
| bir mikrop tabakası. | ||
| Baskın veya resesif tip | ||
| miras | ||
| Hamartoneoplastik | nörofibromatozis | |
| sendrom: | İki veya üç germ hücresi tutulur | Recklinghausen |
| yaprak; | ||
| neoplazi eğilimi; | ||
| genellikle baskın kalıtım şekli | ||
| Malformasyon sendromu veya | Down Sendromu, | |
| bozulmalar (organlar) | Birinde iki veya daha fazla gelişimsel kusur | TAR sendromu, |
| yeni doğan. karakterize | alkol sendromu | |
| embriyonik pleiotropi. | cenin | |
| Biyokimyasal doğrulama | ||
| imkansız veya nadir. etiyoloji | ||
| farklı - monogenik, kromozomal | ||
| veya teratojenik | ||
| Deformite sendromu (alan | ||
| gövde) | Form veya pozisyon ihlali | Potter sendromu |
| başlangıçta normal olarak oluşturulmuş | ||
| vücudun organları veya bölümleri. | ||
| Vakaların çoğu açıklanıyor | ||
| ihlal motor aktivitesi | ||
| fetüs (hipokinezi). | ||
| Kas-iskelet sistemi genellikle etkilenir. | ||
| sistem. |
Doğum kusuru, yeni doğmuş bir bebeğin vücut yapısındaki veya kimyasındaki bir anormalliktir. Bu neden olabilir kalıtsal faktörler (genetik nedenler), etkinin bir sonucu olarak çevre rahimdeki embriyo veya fetüsü veya faktörlerin bir kombinasyonunu etkileyen. Çoğu zaman, doğum kusurlarının nedenleri belirlenemez.
Doğum kusurları bazen konjenital anomaliler olarak adlandırılır. Doğumda mevcut olan anomaliler, hastalık, fiziksel veya zihinsel engellilikle sonuçlanmadıkça genellikle doğum kusuru olarak kabul edilmez. Örneğin, benler nadiren bir doğum kusuru olarak kabul edilir çünkü genellikle sağlık sorunlarına neden olmazlar.
Çocukların yüzde 3 ila 5'inin bir tür doğum kusuruna sahip olduğu tahmin edilmektedir. Bazı doğum kusurları nadiren ortaya çıkar. Bazı doğuştan kalp kusurları gibi diğerleri daha yaygındır.
Kalıtsal faktörler ve doğum kusurları
Her birimizin ebeveynlerimizden miras kalan genleri var. Genler, doğuştan gelen özelliklerimizi veya özelliklerimizi belirler. Doğum kusurları durumunda, genler de anormallikleri etkileyebilir.
Bir çocuk, biri anneden diğeri babadan olmak üzere her genin iki kopyasını miras alır. Kusurlu gen baskınsa, verilen kopyayı miras alan çocuk kusurlu olacaktır. Bunun nedeni, kusurlu kopyanın diğer ebeveynden miras alınan normal kopyaya "baskın" olmasıdır. Ancak kusurlu gen resesif ise, çocuk biri anneden diğeri babadan olmak üzere iki kusurlu kopya miras alacaktır.
Otozomal dominant doğum kusurlarının örnekleri, bir sinir sistemi bozukluğu olan Huntington hastalığı, uzamış kemikler ve kalp problemleriyle karakterize olan Marfan sendromudur. Huntington hastalığı gibi bazı doğuştan hastalıklar uzun yıllar belirti göstermez.
Diğer doğum kusurları, X kromozomundaki genler tarafından belirlenir (X ve Y kromozomları bebeğin cinsiyetini belirler). Hemofili, doğuştan kan hastalıkları ve renk körlüğü, X'e bağlı doğum kusurlarına örnektir.
Bununla birlikte, birçok kalıtsal doğum kusuru, basitçe baskın, resesif veya X'e bağlı değildir; çoklu kusurlu genler de ortaya çıkar.
Kromozom anormallikleri
Bazı doğum kusurlarına fazladan, eksik, eksik veya hatalı biçimlendirilmiş kromozomlar neden olur. En yaygın doğum kusurlarından biri olan Down sendromu, genellikle hücrelerde fazladan bir kromozom bulunmasından kaynaklanır. Down sendromu zeka geriliği, boy kısalığı ve işaretler yüzler. Cinsiyet kromozomlarıyla ilişkili kusurlar, kısırlık (çocuk sahibi olamama) dahil olmak üzere cinsel gelişimde sorunlara yol açabilir.
Çevresel faktörler
Doğum kusurları çevresel faktörlerden de kaynaklanabilir. Buradaki çevre, annenin vücudunu ifade ediyor, ancak bilim adamları üzerinde çalışıyorlar. olası etki doğum kusurlarının oluşumu ve Dünya'nın çevresel koşulları üzerine.
 Aşırı tüketen hamileler erken aşamalar hamilelik var artan risk fetüsü olan çocukları doğurmak alkol sendromu. onunla çocuklar doğuştan hastalık büyüme, görünüm ve çeşitli kusurlara sahip olabilir. zihinsel kapasite. Ve ölçülü alkol tüketimi bile fetüse zarar verebilir. Hamilelik sırasında sigara içmek, bebeğin daha düşük doğum ağırlığına sahip olma olasılığını artırır ve diğer doğum kusurları riskini artırır.
Aşırı tüketen hamileler erken aşamalar hamilelik var artan risk fetüsü olan çocukları doğurmak alkol sendromu. onunla çocuklar doğuştan hastalık büyüme, görünüm ve çeşitli kusurlara sahip olabilir. zihinsel kapasite. Ve ölçülü alkol tüketimi bile fetüse zarar verebilir. Hamilelik sırasında sigara içmek, bebeğin daha düşük doğum ağırlığına sahip olma olasılığını artırır ve diğer doğum kusurları riskini artırır.
Örneğin hamile kadınlardaki bazı hastalıklar bebekte sağırlığa, körlüğe ve doğumsal kalp kusurlarına neden olabilir. Zührevi hastalıklar fetüse veya yenidoğana geçebilir.
Bazı ilaçlar doğum kusurları ile ilişkilidir. En iyi bilineni talidomiddir, yatıştırıcı. Sakinleştiriciler, antibakteriyeller ve diğer birçok ilaç antikanser ilaçları doğumsal anomalilere neden olabilir.
Başka çevresel faktörler doğum kusurları riskini artırdığı düşünülen aşağıdakileri içerir: zayıf beslenme ve annenin yaşı. Örneğin, hamile bir kadın ne kadar yaşlıysa, Down sendromlu bir çocuk doğurma olasılığı o kadar yüksektir.
Doğum kusurlarının teşhisi
 Bazı doğum kusurları, bebek daha anne karnındayken teşhis edilebilir. Kullanılan prosedür ultrasonik dalgalar ekranda fetüsün görüntüsünü elde etmek, doktorun bazı malformasyonları tespit etmesine yardımcı olabilir. Örneğin, bazı omurga kusurları ultrason kullanılarak tespit edilebilir.
Bazı doğum kusurları, bebek daha anne karnındayken teşhis edilebilir. Kullanılan prosedür ultrasonik dalgalar ekranda fetüsün görüntüsünü elde etmek, doktorun bazı malformasyonları tespit etmesine yardımcı olabilir. Örneğin, bazı omurga kusurları ultrason kullanılarak tespit edilebilir.
Adı verilen bir prosedürde, fetüsü çevreleyen küçük bir sıvı örneği incelenir. Bu test, konjenital metabolik kusurları ve kromozomal anormallikleri tespit etmek için kullanışlıdır.
Birçok doğum kusuru, bir doktorun yeni doğmuş bir bebeğin fiziksel muayenesi ile teşhis edilebilir. dahil olmak üzere diğer testler röntgen doktorlar doğum kusurlarından şüpheleniyorsa kullanılabilir. Kan testleri, kan veya vücut kimyasındaki belirli anormallikleri tespit edebilir.
Doğum kusurlarının tedavisi
Her doğum kusuru, buna sahip olan kişinin yaşam kalitesini etkilemez. Bazı doğum kusurlarının, görünüş dışında çok az etkisi vardır.
Bazı doğum kusurları, onları önlemek veya azaltmak için tedavi edilebilir. Zararlı etki. Cerrahi, çarpık ayak, yarık damak gibi doğuştan malformasyonları düzeltmek için ameliyatlar yapabilir. Yarık dudak, yapısal ve gastrointestinal sistem. Tedavi kistik fibrozis semptomlarını azaltabilir, kalıtsal hastalık nefes alma ile ilişkilidir. Bazı durumlarda hidrosefali gibi bozukluklar doğumdan önce düzeltilebilir.
 Doktorlar bazen tedavi edebilir doğuştan bozukluklar ilaçlar yoluyla vücut kimyası ve özel diyetler. Örneğin, hızlı tedavi ciddi sonuçlara yol açabilecek bir metabolik bozukluk olan fenilketonüride beyin hasarını önleyebilir. zeka geriliği. Özel Eğitim, rehabilitasyonun yanı sıra özel cihaz ve cihazların kullanılması, zihinsel ve zihinsel bozuklukların bir kısmını telafi etmeye yardımcı olabilir. fiziksel engeller körlük ve sağırlık gibi.
Doktorlar bazen tedavi edebilir doğuştan bozukluklar ilaçlar yoluyla vücut kimyası ve özel diyetler. Örneğin, hızlı tedavi ciddi sonuçlara yol açabilecek bir metabolik bozukluk olan fenilketonüride beyin hasarını önleyebilir. zeka geriliği. Özel Eğitim, rehabilitasyonun yanı sıra özel cihaz ve cihazların kullanılması, zihinsel ve zihinsel bozuklukların bir kısmını telafi etmeye yardımcı olabilir. fiziksel engeller körlük ve sağırlık gibi.
Doğum kusurlarının önlenmesi
Hiç kimse bir çocuğun "kusursuz" ve sağlıklı doğacağını garanti edemez. Bununla birlikte, bir bebeğin doğum kusurlarına sahip olma şansını en aza indirmenin yolları vardır.
Hamile kadınlar sigara içmemeli veya kullanmamalıdır. alkollü içecekler doktor tarafından reçete edilmedikçe hiçbir ilaç almamalıdırlar. Bazı vitaminler uygun miktarlarda alındığında bazı doğum kusurlarını önlemeye yardımcı olabilir. Örneğin, folik asit hamilelik sırasında bazı spinal ve merkezi sinir sistemi kusurlarını önlemeye yardımcı olabilir. Hamilelikten çok önce aşılama, hamilelik sırasındaki hastalıklardan kaynaklanabilecek doğum kusurlarını önleyebilir.
Sorumluluk reddi: Bu makalede doğum kusurları hakkında verilen bilgiler yalnızca okuyucuyu bilgilendirme amaçlıdır. Bir sağlık profesyonelinin tavsiyesinin yerine geçemez.
