Urođene mane i anomalije. Urođene mane: uzroci, prevencija i liječenje
Neki od dolje prikazanih urođenih mana prilično su poznati, dok su drugi iznimno rijetki. Ali kako god bilo, svi su prilično jezivi, misteriozni i tragični. Tako…
Ovo je rijetko stanje (otprilike jedno na 200 000 poroda) u kojem se blizanci rađaju spojeni u jednom ili više dijelova tijela. U 70-75% svih slučajeva, sijamski blizanci su ženke. Otprilike polovica su mrtvorođena. Ponekad se mogu odvojiti, što dopušta sijamski blizanci uživo puni život, ali najčešće je to nemoguće.
Hipertrihoza (Ambramsov sindrom)
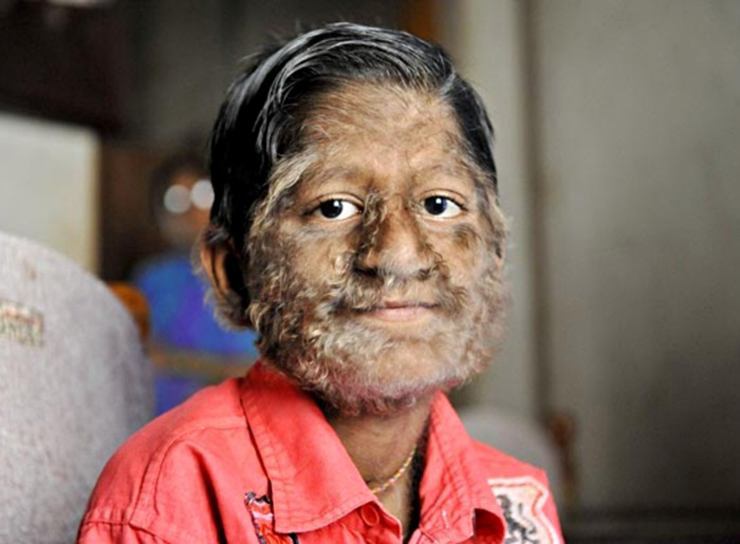
Hipertrihoza je bolest koja se manifestira u višak rasta kosa neobična za ovo područje kože. Srećom, to je vrlo važno, a trenutno samo 40 ljudi u svijetu boluje od hipertrihoze. Bolest je izuzetno iscrpljujuća za djecu jer ih vršnjaci često odbacuju.
Sirenomelija (sindrom sirene)
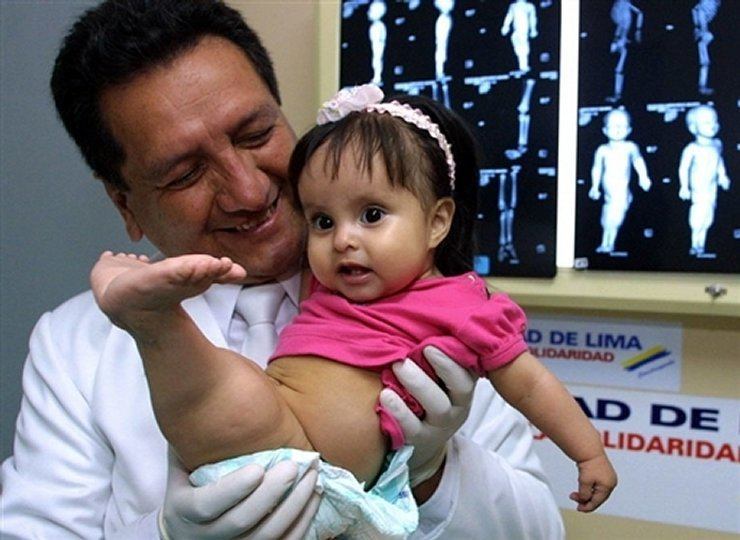
Sirenomelija je razvojna anomalija koja se očituje kao spajanje Donji udovi. Javlja se u jednom slučaju na 100 tisuća novorođenčadi. U pravilu dovodi do smrti 1-2 dana nakon rođenja, to je zbog čudnog razvoja i funkcioniranja bubrega i Mjehur. Međutim, postoje slučajevi kada djeca s ovom anomalijom (čak i bez kirurška intervencija) živio je nekoliko godina. Tako je američka djevojčica Shiloh Pepin, koja je patila od sirenomelije, uspjela živjeti više od 10 godina.
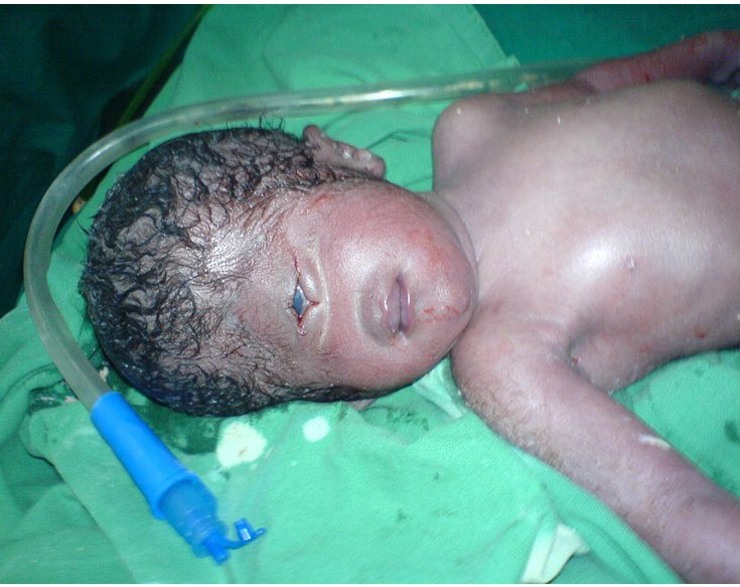
Cyclopia je, kao što možda pretpostavljate, dobila ime po poznatom mitskom biću Kiklopu. Djeca rođena s ciklopijom imaju samo jedno oko, smješteno u sredini glave. U 100% svih slučajeva novorođenčad umire u prvim danima života.
Vrsta blizanačke fuzije u kojoj glava normalno dijete glava blizanca urasta bez tijela. Povijest poznaje samo deset zabilježenih primjera ove anomalije, a samo u tri od njih dijete je ostalo živo nakon rođenja. U jednom slučaju, druga se glava mogla nasmiješiti, treptati, plakati i sisati majčinu dojku.
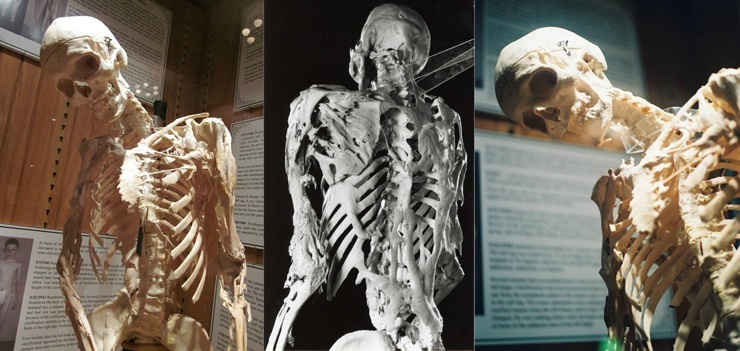
Rijetka bolest(1 slučaj od 2 milijuna), nastaje kao rezultat mutacije gena i manifestira se urođenim razvojnim defektima - prvenstveno zakrivljenima palčevi zaustavljanje i poremećaji u vratne kralježnice kralježnice. Osnova fibrodisplazije je formacija upalni procesi u tetivama, ligamentima, fascijama, aponeurozama i mišićima, koji u krajnji rezultat dovodi do njihove kalcifikacije i okoštavanja. Bolest se također naziva "bolest drugog kostura", jer zapravo tamo gdje su potrebni normalni protuupalni procesi u tijelu, počinje rast kostiju.
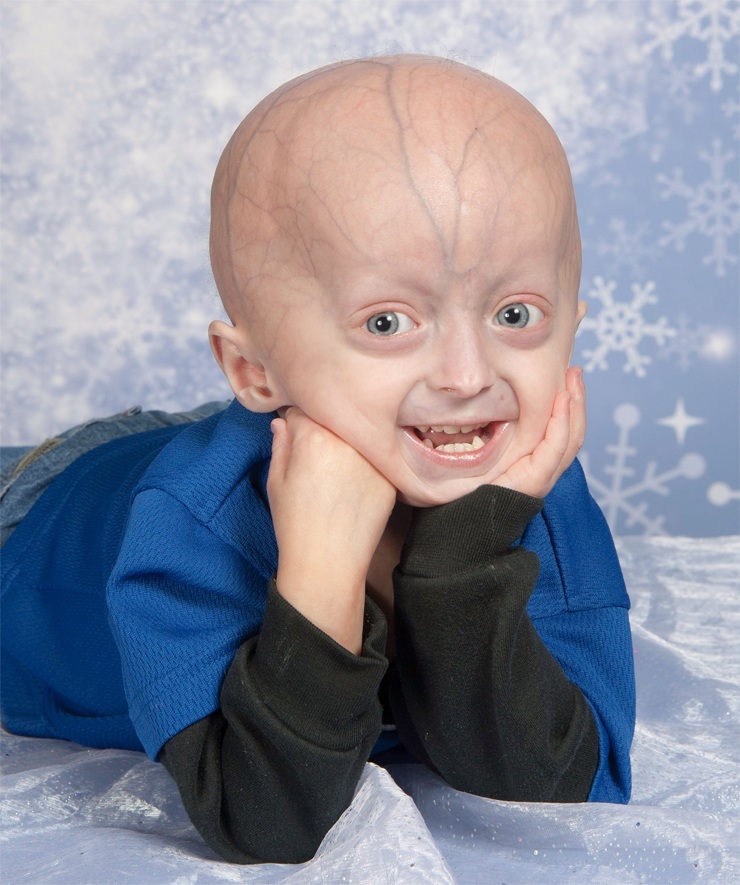
Progerija - najrjeđa genetski defekt, kod kojih se javljaju kožne promjene i unutarnji organi, uvjetovano rano starenje tijelo. U svijetu nije zabilježeno više od 80 slučajeva progerije.
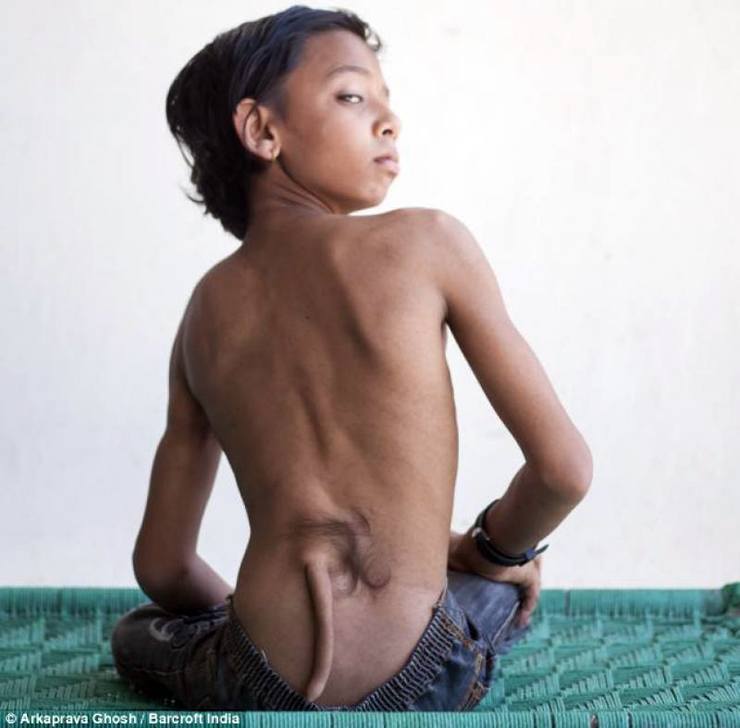
Urođena mana, u kojem se beba rađa s polufunkcionalnim repom s mišićima, živcima, kožom i krvne žile. Vjeruje se da bi to moglo biti uzrokovano mutacijom gena.

Anencefalija – potpuna ili djelomična odsutnost moždane hemisfere mozak, kalvarijske kosti i meka tkiva. Javlja se otprilike jednom u 10 tisuća novorođenčadi (u SAD-u), češće u ženskih fetusa. Defekt je fatalan u 100% slučajeva. 50% fetusa s anencefalijom umire u maternici, preostalih 50% se rađaju živi, ali samo 66% može preživjeti nekoliko sati (međutim, poznato je da neki žive oko tjedan dana). Stephanie Keene, poznatija pod nadimkom Baby Kay, smatra se "najdugovječnijom" među anencefaličarima, koja je s ovom strašnom dijagnozom živjela 2 godine i 174 dana.
Podijelite na društvenim mrežama mreže
Otprilike 2-3% novorođenčadi ima ozbiljne kongenitalne malformacije. Embriološki, takvi se nedostaci klasificiraju u tri glavne klase (tablica 36-6):
Urođene mane kao rezultat nepotpune morfogeneze;
Urođene mane koje su rezultat ponavljajuće morfogeneze;
Urođene mane koje su rezultat aberantne morfogeneze. Nepotpuna morfogeneza je najčešća patologija, aberantna - najrjeđa.
Tablica 36-6.
(Cohen M.M., 1997.)
Gotovo sve urođene malformacije javljaju se u embrionalno razdoblje(3-10. tjedan gestacije), u razdoblju kada dolazi do diferencijacije organa (Tablica 36-7).
Urođene mane mogu biti jednostavne i složene. Kako kasniji datum pojava urođene mane, vjerojatnije da neće biti patogenetski povezanih defekata u obližnjim embrionalnim strukturama (jednostavne kongenitalne malformacije). Ako se defekt pojavi u ranim fazama embriogeneze, vjerojatnost zahvaćanja obližnjih struktura je prilično visoka, a kaskada višestrukih urođene mane razvoja ili slijeda (slijeda) kongenitalnih malformacija. Primjer je sekvenca Pierre Robin, kada je primarni defekt intrauterina hipoplazija Donja čeljust uzrokuje poremećaj u procesu retrakcije jezika, što zauzvrat dovodi do rascjepa nepca.
Tablica 36-7. Vrijeme nastanka kongenitalnih malformacija (Cohen M.M., 1997.)
Kongenitalna malformacija u svom klinička manifestacija mogu biti minimalno (bifurkacija uvule) i maksimalno izraženi (rascjep nepca). U slučaju minimalne manifestacije, definirat će se kao manja anomalija razvoj. Složena malformacija ili sekvenca također se može pojaviti s minimalnim klinička verzija.
Na primjer, slijed holoprozencefalije u svojoj najtežoj inačici karakterizira kongenitalna malformacija moždanih hemisfera i anomalije lica - odsutnost nosnih struktura, hipotelorizam, premaksilarna agenezija s rascjepom usne i alveolarni nastavak Gornja čeljust; u svom minimalnom kliničkom obliku, karakteriziran je kombinacijom hipotelorizma s jednim maksilarnim sjekutićem. Poznavanje toga iznimno je važno za obiteljsku genetsku prognozu djeteta s holoprozencefalijom – pregled roditelja na minimalne kliničke manifestacije.
DEFORMACIJA
Ova vrsta urođene mane pojavljuje se u otprilike 1-2% novorođenčadi. Najčešći defekti su klupavo stopalo, urođeno iščašenje kuka i posturalna skolioza. Deformacije najčešće nastaju u kasnom fetalnom razdoblju kao posljedica utjecaja triju glavnih uzroka i predisponirajućih čimbenika (tablica 36-8): mehaničkih uzroka; kongenitalne malformacije; funkcionalni razlozi.
Mehanički uzroci deformacija su najčešći i javljaju se u pozadini fetalne hipokinezije. U istraživanju na 4.500 novorođenčadi pokazalo se da je od sve novorođenčadi s deformitetima 1/3 djece imala dva ili više deformiteta. Ovaj niz urođene deformacije dobro ilustrira primjer gdje je rigidnost maternice uzrok triju deformiteta - plagiocefalije, asimetrije donje čeljusti i klupavosti u jednog novorođenčeta.
Tablica 36-8. Predisponirajući čimbenici u razvoju deformiteta (Cohen M.M., 1997.)
| Mehanički | |
| uzroci | Rigidnost maternice i trbušnih mišića (u kombinaciji s |
| prvi porod) | |
| Nizak rast i smanjena tjelesna veličina kod trudnice | |
| žene | |
| Hipoplazija karlični prsten | |
| Hipoplazija maternice | |
| Dvoroga maternica | |
| Leiomiom maternice | |
| Neobično mjesto implantacije | |
| Kronično istjecanje amnionske tekućine | |
| Niska voda ( raznih etiologija) | |
| Neobičan položaj fetusa | |
| Rano umetanje glave fetusa u zdjelicu | |
| Višeplodna trudnoća | |
| Kongenitalne malformacije fetusa | |
| Veliko voće(kongenitalna makrosomija) | |
| Makrocefalija ili fetalni hidrocefalus | |
| Urođene mane | Spina bifida; |
| fetalni razvoj kao | |
| uzrok deformacije | Druge kongenitalne malformacije živčani sustav fetus |
| Fetalna bubrežna agenezija (obostrana) | |
| Teška hipoplazija bubrega | |
| Teška policistična bolest bubrega | |
| Atrezija uretre | |
| Funkcionalan | Neurološki poremećaji(kongenitalna hipotenzija) |
| uzroci | |
| Poremećaji mišića | |
| Nedostaci vezivno tkivo | |
Gotovo sve teške urođene mane mokraćni sustav uzrokuju oligohidramnij, koji, pak, uzrokuje Potterov sindrom (neobično lice fetusa, višestruke kontrakture udova).
Funkcionalni uzroci deformiteta uključuju raznih oblika kongenitalna hipotonija novorođenčadi i neuromuskularni tipovi artrogripoze. Kongenitalna hipotonija može se kombinirati s mikrognatijom, mikroglosijom i istaknutim bočnim šavovima tvrdo nepce, abnormalna fleksija šake i stopala, ravni valgus stopala i drugih deformiteta. Artrogripozu karakterizira kongenitalna ukočenost udova i fiksacija zglobova u karakterističnom položaju.
POREMEĆAJ
Točna učestalost poremećaja je nepoznata, otkriva se u 1-2% novorođenčadi. Prvi istraživač koji je opisao ovaj tip patologije u monografiji iz 1968. “Fetal Malformations Caused by Amnion Rupture During Gestation”, bio je R. Torpin (Cohen M.M., 1997.). Smetnje nastaju kao posljedica izloženosti razni razlozi: vaskularni faktori, anoksija, infekcije, zračenje, teratogeni, amnionske vrpce, mehanički faktori. Vrsta i težina poremećaja ovise o trajanju trudnoće, mjestu izloženosti i stupnju oštećenja tkiva. Najčešće se poremećaji javljaju tijekom fetalnog razdoblja, ali teratogeni učinci karakteristični su za embrionalnu morfogenezu. Neki od ovih učinaka koji se javljaju tijekom embrionalnog razdoblja "fenokopiraju" kongenitalne malformacije. Na primjer, amnionske vrpce rani datum Trudnoća može uzrokovati anencefaliju, rascjep usne i nepca i redukcijske defekte udova. Najteže diferencijalna dijagnoza između kongenitalne malformacije i poremećaja kao posljedice vaskularne patologije (Tablica 36-9).
Tablica 36-9. Mehanizmi vaskularnog poremećaja u embriju i fetusu (Cohen M.M., 1997.)
| Patogeneza | Strukturna anomalija |
| Uništavanje embrionalnog kapilarna mreža | Rani amnionski slijed, kompleks ud-torzo stijenka, redukcijske anomalije udova, hipoplazija maksilarne regije i udova |
| Postojanost embrionalnih žila | Defekti ekstremiteta: radijalna aplazija, aplazija tibije, aplazija fibule, klupavo stopalo |
| Prijevremena amputacija embrionalnih žila | Slijed patologije potključna arterija(Poljska, Mobius, Klippel-Feil sekvence), gastroshiza, potkovičasti bubreg |
| Poremećeno vaskularno sazrijevanje | Kapilarni hemangiomi, arteriovenske fistule, aneurizme (Berry aneurizme) |
| Okluzija (vanjska kompresija) krvnih žila | Anomalije povezane s leiomiomima, sa tubarna trudnoća i dvoroga maternica |
| Okluzija (embolička tromboza) krvnih žila | Porencefalija, hidranencefalija, mikrocefalija, atrezija žučnog mjehura, distalna sindaktilija, hemifacijalna mikrosomija (rijetko), bilateralni anorhizam, kožna aplazija |
| Hemodinamski poremećaj | Abnormalnosti uzrokovane korištenjem kokaina tijekom trudnoće |
Izolirani kongenitalni nedostaci u razvoju ne uzrokuju poteškoće u dijagnozi. Sasvim drugačija situacija je u području višestrukih prirođenih mana u razvoju, gdje su empirijska iskustva i znanja o dijagnostici i liječenju djece s izoliranim prirođenim greškama ne samo nedostatna, nego često i pogrešna.
Potrebe kliničke prakse pridonijele su proširenju istraživanja etiologije i patogeneze višestrukih prirođenih mana u razvoju. Taj dio kasnije je nazvan sindromologija. Jedna od jasnih ilustracija razlike između sindromologije i klasične medicine, tj. medicine, gdje se dijagnosticira i proučava izolirana patologija jednog organa ili sustava, jest činjenica da in klasična medicina Tijekom 20. stoljeća opisano je samo nekoliko novih bolesti ( radijacijske bolesti, legionarska bolest, AIDS, lajmska bolest), dok je u sindromologiji broj takvih nozoloških oblika premašio 5000, a godišnje se opiše najmanje 80 novih.
Za neke oblike sindromne patologije moderna molekularna genetika omogućila je lokalizaciju gena koji ih određuju i proučavanje produkata transkripcije gena, koji su često predstavljeni membranskim receptorima ili faktorima rasta tkiva. Na primjer, kod Hirschsprungove bolesti identificirane su dvije mutacije različitih gena: RET onkogen i endotelin B receptor, što je omogućilo razlikovanje dva genetska tipa ove kongenitalne patologije.
Sindromologija je iznimno široko područje koje pokriva gotovo sve specijalnosti medicine. Otprilike 1% svih novorođenčadi ima višeplod kongenitalne anomalije ili sindrome. Od toga je danas u 40% slučajeva moguće dijagnosticirati određeni sindrom, a preostalih 60% zahtijeva njihovu identifikaciju kao „nove“ sindrome. Iako su mnogi sindromi prilično rijetki, ukupni sindromski oblici patologije čine kvantitativno značajan dio medicine.
Tablica 36-10. Mehanizmi vaskularne destrukcije u embriju i fetusu (Cohen M.M., 1997.)
| Okolišni čimbenici | Teratogeni učinci |
| Farmakološki lijekovi | |
| Talidomid | Redukcija anomalija udova. Hipoplazija pojasa gornjih ekstremiteta. |
| Abnormalnosti uha | |
| Alkohol | Odgoditi tjelesni razvoj. Neobičan fenotip (kratke oči |
| pukotine). Mikrocefalija, mentalna retardacija | |
| DietStyleOestrol | Adenomatoza rodnice. Cervikalna erozija. Vaginalni adenokarcinom |
| (rijetko) | |
| Varfarin | |
| Hipoplazija nosne hrskavice. Kongenitalne mane središnjeg živčanog sustava. Tačkasta kalcifikacija | |
| epifize | |
| hidantoin (dilantin) | |
| Zakašnjeli fizički razvoj. Neobičan fenotip. Mikrocefalija, mentalna | |
| zaostalost | |
| Trirmetadion | |
| Zakašnjeli psihomotorni razvoj. Neobičan fenotip (lučni | |
| obrve). Rascjep usne ili nepca | |
| Aminopterin | |
| Metotreksat | Spontani pobačaji. Kongenitalni hidrocefalus. Zakašnjeli fizički razvoj |
| Tia. Neobičan fenotip | |
| Streptomicin | |
| Kongenitalni senzorineuralni gubitak sluha | |
| tetraciklin | |
| Kongenitalna hipoplazija zubna caklina. Bojenje zuba (žuti zubi) | |
| Natrijev valproat | |
| Defekti neuralne cijevi (Spina bifida) | |
| retinol | |
| Spontani pobačaji. Kraniofacijalne anomalije. Defekti neuralne cijevi | |
| Pripravci litija | |
| Urođene srčane mane (Ebsteinova anomalija) | |
| Antitiroidni lijekovi | |
| V G. Gušavost | |
| Androgeni i visoke doze | |
| maskulinizirajući | maskulinizacija |
| progestini | |
| Penicilamin | |
| Hiperelastična koža. Kongenitalna patologija vezivno tkivo | |
| Metil živa (živa) | Kemijske tvari |
| Kongenitalna atrofija mozak. Spastičnost, konvulzije. Mentalno | |
| zaostalost | |
| voditi | |
| Zakašnjeli fizički razvoj. Neuobičajena boja kože (siva boja) | |
| Pušenje | Fizički, prehrambeni i drugi učinci |
| Spontani pobačaji. IUGR | |
| Ionizirana radiacija | |
| Šteta ovisi o stupnju trudnoće. Spontani pobačaji. Kongenitalna | |
| nedostaci u razvoju (18-36 dana trudnoće). Mikrocefalija i mentalna retardacija | |
| Gubitak (8-15. tjedan trudnoće) | |
| Hipertermija | |
| Kongenitalne mane središnjeg živčanog sustava | |
| Dijabetes melitus majke | |
| UPS. Sindrom kaudalne regresije | |
| Nedostatak joda u hrani | |
| PKU kod majke | Gušavost. Mentalna retardacija i odgođeni fizički razvoj |
| Pobačaj, mikrocefalija, mentalna retardacija |
Ako su simptomi ili kompleks simptoma uzrokovani jednim uzrokom (monokauzalna etiologija), tada pojam sindrom označava nozološki oblik bolesti (nozološki sindrom) i u tom je smislu sinonim za pojam "bolest". U praksi, prema preporukama međunarodnih stručnjaka, termin "bolest" bolje je koristiti u slučajevima s progresivnim tijekom bolesti.
Dakle, sindrom je etiološki određena bolest s pleiotropnim (višestrukim) učinkom.
Primjer takvog sindroma je povijest Recklinghausenove bolesti ili neurofibromatoze tipa 1. Godine 1849. Robert Smith, vodeći kirurg na Medicinskom fakultetu u Dublinu, objavio je kliničke i patološke značajke dvaju slučajeva neurofibromatoze i naveo podatke iz 75 ranijih publikacija u medicinske literature. Međutim, samo u izvješću Recklinghausena (von Recklinghausen, 1882) potkrijepljena je ideja o neurofibromatozi kao nosološkom obliku. Sada se pokazalo da je ova patologija jedna od najčešćih nasljednih bolesti ljudi i da se javlja s učestalošću od 1 u 2000 rođenih. Moderno dijagnostički kriteriji ove bolesti na temelju takvog karakteristični simptomi, poput hiperpigmentacije kože (tip cafe au lait), kongenitalnih lažnih zglobova ili zakrivljenosti kostiju donjih ekstremiteta, razvili su se tek 1987. godine. Treba napomenuti da je dijagnoza moguća u slučajevima kada pacijent ima dvije od sljedeće znakove i pod uvjetom da nisu simptomi neke druge bolesti.
Dijagnostički kriteriji za neurofibromatozu tipa 1 (Recklinghausenova bolest) (SZO memorandum, 1992.):
Tijekom pregleda pod umjetnim svjetlom pacijenta koji nije dosegao pubertet, otkriva se najmanje pet svijetlosmeđih pigmentnih mrlja (više od 5 mm na najširem mjestu); pri pregledu pacijenta koji je dosegao pubertet - najmanje 6 pigmentnih mrlja (više od 15 mm na najširem mjestu);
Prisutnost, prema povijesti bolesti ili klinički pregled, dva ili više neurofibroma bilo kojeg tipa ili jedan pleksiformni neurofibrom;
Višestruki, poput pjega tamne mrlje u aksilarnim ili preponskim područjima;
Displazija krila sfenoidalna kost ili kongenitalna zakrivljenost ili stanjivanje dugih kostiju s formiranjem lažni zglob ili bez njega;
Glioma optičkog živca;
Dvije ili više Picheovih točaka/kvržica otkrivenih na šarenici pregledom procjepnom svjetiljkom;
Prisutnost neurofibromatoze tipa 1, prema gore navedenim kriterijima, kod srodnika u prvom koljenu (roditelj, brat ili sestra ili potomak).
Pravovremena dijagnoza Recklinghausenove neurofibromatoze zahtijeva dinamičko promatranje uz periodične CT pretrage glave i leđna moždina s ciljem rana dijagnoza neoplazija središnjeg živčanog sustava.
Već 60-70-ih godina XX. stoljeća. većina kromosomskih i teratogenih sindroma je opisana i veliki iznos genski sindromi. Do početka 80-ih godina XX. stoljeća. Iskustvo je omogućilo ne samo da se razvije jedinstvena međunarodna terminologija, već i da se predlože neki metodološki pristupi za identificiranje "novih" sindroma i proučavanje patogeneze višestrukih razvojnih nedostataka (Spranger J. et al, 1982; Cohen MM., 1982; Cohen MM. , 1997).
Sindromski oblici patologije u praksi također uključuju slučajeve kada dijete, uz bilo koju urođenu manu, ima neobičan fenotip, odnosno prisutnost tri ili više manjih razvojnih anomalija.
Manje razvojne anomalije ili stigme disembriogeneze abnormalne su varijante morfologije pojedini organi ili tkanine koje nemaju medicinsku vrijednost, tj. ne zahtijevaju liječenje. Pojava ovih varijanti povezana je s embrionalnim ili, rjeđe, s plodno razdoblje ljudska morfogeneza. U klinička genetika i sindromologije, manje razvojne anomalije izuzetno su važan dijagnostički znak koji ukazuje na visoku vjerojatnost ozbiljne povrede morfogeneze u obliku kongenitalnih malformacija koje zahtijevaju specijalna dijagnostika a često i naknadne kirurške intervencije (tablica 36-11).
U ljudi je opisano više od 200 informativnih morfogenetskih varijanti klinička praksa Obično se ne koristi više od 80 manjih razvojnih anomalija.
Manje razvojne anomalije novorođenčadi:
glava:
V nenormalan obrazac rasta kose;
V spljošten zatiljak;
V “tuberkuloze” svoda lubanje.
Orbitalno područje:
V epikantski nabori;
V epikantus naličje;
V mongoloidni oblik oka;
V antimongoloidni dio oka;
V kratke palpebralne fisure;
V distopija vanjskih kutova oka;
V hipotelorizam je umjeren;
V umjereni hipertelorizam;
Ptoza je blaga;
V heterokromija šarenica;
V mikrokorneja.
uši:
V primitivni oblik;
V Darwinov tuberkul;
V abnormalni oblik kovrče;
V asimetrične uši;
V rotirane uši;
V smanjene ušne školjke;
V stršeće uši;
V odsutnost tragusa;
Rascjep režnja;
V odsutnost režnja;
V aurikularne jame;
V ušne "izbočine";
V suženje vanjskog zvukovoda.
Nos i filter:
¦¦¦ hipoplazija krila nosa;
¦¦¦ raširene nosnice;
¦¦¦ spljošteni filter;
¦¦¦ filter koji strši.
Područje usta i usne šupljine:
¦¦¦ mikrogenija;
¦¦¦ rascjep jezika;
¦¦¦ aberantni frenulum predvorja usta;
¦¦¦ neonatalni zubi-filter.
¦¦¦ krilati vrat - umjeren;
¦¦¦ fistule na vratu.
Vestigijalna polidaktilija; jedan pregib dlana; abnormalni dermatoglifi; klinodaktilija malih prstiju; skraćivanje 4-5 prstiju; hipoplazija završnih falangi.
Sindaktilija P-Sh prsti; prorezi u obliku sandala; kratki 1. prst; prstohvat (IV-V); zadebljali nokti.
Koža:
Hemangiomi;
Hiperpigmentacija kože i nevusi; Mongoloidne mrlje (kod bijele rase); depigmentacija kože; dodatne bradavice ili areole.
Torzo:
¦¦¦ dijastaza rektusa abdominisa;
¦¦¦ umjerena hipospadija (glave);
¦¦¦ duboki utisci križne kosti.
Kostur:
¦¦¦ udubljenje ili izbočenje prsne kosti.
Područje glave, vrata i ruku najinformativnije je za ove znakove, više od 70% svih manjih razvojnih anomalija nalazi se u ovom području. Dijagnostička vrijednost razlikuju se manje razvojne anomalije. Temeljno je važno da praktični značaj ovih znakova leži u dijagnozi tri ili više anomalija. Svako novorođenče s tri ili više lakših malformacija ima visoku vjerojatnost (90%) ozbiljne kongenitalne malformacije mozga, srca, bubrega ili kralježnice, osim velika vjerojatnost(50%) dijagnoza sindromskog oblika patologije. Ako dijete sa zaostatkom u psihomotornom razvoju ima tri ili više lakših anomalija u razvoju, navodi se visokog rizika organska oštećenja SŽS (vidi tablicu 36-11). Ponekad je samo prisutnost dviju manjih razvojnih anomalija informativna za dijagnozu. Na primjer, hipotelorizam (usko smješten očne jabučice) i jedini gornji sjekutić ukazuju na urođeni defekt mozga tipa prozencefalne skupine.
Dijagnoza urođene razvojne mane djeteta postavlja neonatologu sljedeća pitanja:
Kojoj vrsti patologije pripada ovaj defekt (kongenitalna malformacija, disrupcija, deformacija ili displazija)?
Koliko je često ova urođena mana povezana s drugim urođenim manama ili bolestima koje još nisu klinički vidljive?
Koliko često je ova urođena mana simptom sindromskog oblika patologije?
Koji su sindromi najčešći uz ovu urođenu manu?
Odgovori na ova pitanja su prvi dijagnostički stadij praktični suradnja neonatolog i genetičar. Konačni cilj Ova faza uključuje dijagnozu dodatnih kongenitalnih razvojnih mana ili dijagnozu specifičnog sindroma. U slučaju dijagnosticiranja sindromskog oblika patologije, u većini slučajeva, dalje medicinske taktike glede konzervativnog ili kirurško liječenje te medicinska i genetska prognoza u obitelji bolesnog djeteta. Informacije o prognozi za život i zdravlje s određenim sindromom ima veliki značaj i glavni je cilj medicinskog rada.
Sindromi se mogu analizirati na različitim razinama biološke organizacije:
Na razini poremećaja unutar metaboličkih procesa (dismetabolički sindromi);
Na razini poremećaja tkiva (sindromi displazije);
Na razini morfoloških poremećaja organa (sindromi kongenitalnih malformacija i disrupcijski sindromi);
Na razini poremećaja određenog područja tijela (sindromi deformacije). Sve četiri razine kršenja imaju i praktični značaj, budući da sva četiri biološka tipa sindroma imaju jasne dijagnostičke i genetske kriterije za dijagnozu i prognozu zdravlja (Tablica 36-12).
Dijagnoza sindromskih oblika patologije u novorođenčadi uključuje:
Prilika točna dijagnoza popratne bolesti dijete s višestrukim nedostacima;
Prognoza komplikacija karakterističnih za svaki sindrom tijekom kirurškog odn konzervativno liječenje urođene mane ili bolesti;
Točna procjena mogućnosti kirurškog ili konzervativnog liječenja bolesti (vrijeme i obim kirurškog zahvata, dugoročni rezultati liječenje);
Točna medicinska i genetska prognoza u obitelji.
Ove zaključke dobro ilustriraju sljedeća opažanja iz kliničke prakse.
Tablica 36-12. Biološki tipovi sindromi (Cohen M.M., 1982.)
| Vrsta sindroma (stupanj oštećenja) | Karakteristični znakovi | Primjeri |
| Dismetabolički sindrom (metabolizam) Sindrom displazije (tkiva) | Novorođenčad ima normalan fenotip s progradijentnom kliničkom slikom. Klinički znakovi relativno sličan u usporedbi s drugim vrstama sindroma. Ne postoji povezanost s kongenitalnim malformacijama. Moguća je verifikacija primarnog biokemijskog defekta. Autosomno recesivni tip nasljeđivanja Sindrom jednostavne displazije karakterizira samo oštećenje | PKU, Tay-Sachsova bolest, VGS Marfanov sindrom, Ehlers-Danlosov sindrom, ahondroplazija |
| jedan klicin listić. | ||
| Dominantni ili recesivni tip | ||
| nasljedstvo | ||
| Hamartoneoplastični | Neurofibromatoza | |
| sindrom: | Uključene su dvije ili tri germinativne stanice | Recklinghausen |
| list; | ||
| sklonost neoplaziji; | ||
| obično dominantan način nasljeđivanja | ||
| Sindrom razvojne mane ili | Downov sindrom, | |
| korupcija (organi) | Dva ili više razvojnih nedostataka u jednom | TAR sindrom, |
| novorođenče Karakterizira ga | alkoholni sindrom | |
| embrionalna pleiotropija. | fetus | |
| Biokemijska verifikacija | ||
| nemoguće ili rijetko. Etiologija | ||
| različiti - monogeni, kromosomski | ||
| ili teratogen | ||
| Sindrom deformacije (područje | ||
| tijelo) | Povreda oblika ili položaja | Potterov sindrom |
| u početku normalno formirana | ||
| organa ili dijelova tijela. | ||
| Većina slučajeva je objašnjena | ||
| kršenje motorna aktivnost | ||
| fetus (hipokinezija). | ||
| Obično je zahvaćen mišićno-koštani sustav | ||
| sustav. |
Urođena mana je abnormalnost u tjelesnoj strukturi ili kemiji novorođenčeta. Ovo može biti uzrokovano nasljedni faktori (genetski razlozi), kao rezultat izloženosti okoliš, koji utječe na embrij ili fetus u maternici, ili kombinacija čimbenika. Često se uzroci urođenih mana ne mogu utvrditi.
Urođene mane ponekad se nazivaju kongenitalne anomalije. Abnormalnosti koje su prisutne pri rođenju općenito se ne smatraju urođenom manom osim ako rezultiraju bolešću ili fizičkim ili mentalnim nedostatkom. Na primjer, madeži se rijetko smatraju urođenom manom jer obično ne uzrokuju zdravstvene probleme.
Procjenjuje se da 3 do 5 posto djece ima neku vrstu urođene mane. Neke se urođene mane javljaju rijetko. Druge, poput nekih urođenih srčanih mana, češće su.
Nasljedni faktori i urođene mane
Svatko od nas ima gene naslijeđene od roditelja. Geni određuju naše urođene karakteristike ili osobine. U slučaju urođenih mana, geni također mogu utjecati na abnormalnosti.
Dijete nasljeđuje dvije kopije svakog gena, jednu od majke i jednu od oca. Ako je defektni gen dominantan, dijete koje naslijedi tu kopiju imat će defekt. To se događa jer neispravna kopija "dominira" normalnom kopijom naslijeđenom od drugog roditelja. Ali ako je defektni gen recesivan, dijete će naslijediti dvije defektne kopije: jednu od majke i jednu od oca.
Primjeri autosomno dominantnih urođenih mana uključuju Huntingtonovu bolest, poremećaj živčanog sustava i Marfanov sindrom, koji karakteriziraju duge kosti i srčani problemi. Neke kongenitalne bolesti, poput Huntingtonove bolesti, godinama nemaju simptoma.
Druge urođene mane određene su genima na X kromosomu (X i Y kromosomi određuju spol djeteta). Hemofilija, urođeni poremećaji krvi i sljepoća za boje primjeri su X-kromosomskih urođenih mana.
Mnogi nasljedni urođeni defekti, međutim, nisu samo dominantni, recesivni ili X-kromosomski; postoji i više defektnih gena.
Kromosomske abnormalnosti
Neki urođeni defekti uzrokovani su dodatnim, nedostajućim, nepotpunim ili izobličenim kromosomima. Downov sindrom, jedna od najčešćih urođenih mana, obično je uzrokovan prisutnošću viška kromosoma u stanicama. Downov sindrom karakterizira mentalna retardacija, nizak rast i razlikovna obilježja lica. Defekti povezani sa spolnim kromosomima mogu dovesti do problema sa spolnim razvojem, uključujući sterilitet (nemogućnost imati djecu).
Okolišni čimbenici
Urođene mane također mogu biti uzrokovane okolišnim čimbenicima. Okolina ovdje znači majčino tijelo, međutim, znanstvenici proučavaju mogući utjecaj o pojavi urođenih mana i ekološkim uvjetima Zemlje.
 Trudnice koje konzumiraju prekomjerne količine rani stadiji imati trudnoću povećan rizik rađati djecu s fetalnim alkoholni sindrom. Djeca s ovim urođena bolest može imati različite nedostatke u rastu, izgledu i mentalne sposobnosti. Čak i umjerena konzumacija alkohola može uzrokovati štetu fetusu. Pušenje tijekom trudnoće povećava vjerojatnost da će beba imati manju porođajnu težinu i povećava rizik od drugih urođenih mana.
Trudnice koje konzumiraju prekomjerne količine rani stadiji imati trudnoću povećan rizik rađati djecu s fetalnim alkoholni sindrom. Djeca s ovim urođena bolest može imati različite nedostatke u rastu, izgledu i mentalne sposobnosti. Čak i umjerena konzumacija alkohola može uzrokovati štetu fetusu. Pušenje tijekom trudnoće povećava vjerojatnost da će beba imati manju porođajnu težinu i povećava rizik od drugih urođenih mana.
Neke bolesti kod trudnica, primjerice, mogu dovesti do gluhoće, sljepoće i urođenih srčanih mana djeteta. Venerične bolesti mogu se prenijeti na fetus ili novorođenče.
Neki lijekovi povezani su s urođenim manama. Najpoznatiji je talidomid. sedativ. Mnogi drugi lijekovi, uključujući sredstva za smirenje, antibakterijske i antitumorski lijekovi, može uzrokovati urođene abnormalnosti.
ostalo okolišni čimbenici za koje se vjeruje da povećavaju rizik od urođenih mana uključuju loša prehrana i dobi majke. Na primjer, što je trudnica starija, veća je vjerojatnost da će roditi dijete s Downovim sindromom.
Dijagnostika urođenih mana
 Neke urođene mane mogu se dijagnosticirati dok je beba još u maternici. Postupak koji koristi ultrazvučni valovi za dobivanje slike fetusa na ekranu, može pomoći liječniku u otkrivanju nekih malformacija. Na primjer, neki defekti kralježnice mogu se otkriti pomoću ultrazvuka.
Neke urođene mane mogu se dijagnosticirati dok je beba još u maternici. Postupak koji koristi ultrazvučni valovi za dobivanje slike fetusa na ekranu, može pomoći liječniku u otkrivanju nekih malformacija. Na primjer, neki defekti kralježnice mogu se otkriti pomoću ultrazvuka.
U postupku koji se zove , ispituje se mali uzorak tekućine koja okružuje fetus. Ovaj test je koristan za otkrivanje urođenih metaboličkih nedostataka i kromosomskih abnormalnosti.
Mnoge urođene mane mogu se dijagnosticirati liječničkim fizičkim pregledom novorođenčeta. Ostali testovi uključujući X-zrake, može se koristiti ako liječnici sumnjaju na urođene mane. Krvni testovi mogu otkriti određene abnormalnosti u krvi ili tjelesnoj kemiji.
Liječenje urođenih mana
Ne utječe svaka urođena mana na kvalitetu života osobe koja je ima. Neke urođene mane imaju mali učinak, osim možda u izgledu.
Neke urođene mane mogu se liječiti kako bi se spriječile ili smanjile štetni učinci. Kirurgija može izvesti operacije za ispravljanje urođenih malformacija kao što su klupko stopalo, rascjep nepca, napuknuta usna, strukturne i gastrointestinalni trakt. Liječenje može smanjiti simptome cistične fibroze, nasljedna bolest povezana s disanjem. U nekim slučajevima, poremećaji poput hidrocefalusa mogu se ispraviti prije rođenja.
 Liječnici ponekad mogu liječiti urođeni poremećaji tjelesne kemije putem lijekova i posebne dijete. Na primjer, brzo liječenje može spriječiti oštećenje mozga uzrokovano fenilketonurijom, metaboličkim poremećajem koji može dovesti do teških mentalna retardacija. Posebna edukacija, rehabilitacija, kao i korištenje posebnih uređaja i naprava može pomoći u nadoknadi nekih psihičkih i tjelesne nedostatke, kao što su sljepoća i gluhoća.
Liječnici ponekad mogu liječiti urođeni poremećaji tjelesne kemije putem lijekova i posebne dijete. Na primjer, brzo liječenje može spriječiti oštećenje mozga uzrokovano fenilketonurijom, metaboličkim poremećajem koji može dovesti do teških mentalna retardacija. Posebna edukacija, rehabilitacija, kao i korištenje posebnih uređaja i naprava može pomoći u nadoknadi nekih psihičkih i tjelesne nedostatke, kao što su sljepoća i gluhoća.
Prevencija urođenih mana
Nitko ne može jamčiti da će se dijete roditi "savršeno" i zdravo. Međutim, postoje načini da smanjite šanse da vaša beba ima urođene mane.
Trudnice ne smiju pušiti niti koristiti alkoholna pića, ne bi smjeli uzimati nikakve lijekove osim ako ih ne prepiše liječnik. Neki vitamini, ako se uzimaju u odgovarajućim količinama, mogu spriječiti neke urođene mane. Na primjer, folna kiselina tijekom trudnoće može pomoći u sprječavanju nekih oštećenja kralježnice i središnjeg živčanog sustava. Cijepljenje puno prije trudnoće može spriječiti urođene mane koje mogu nastati kao posljedica bolesti tijekom trudnoće.
Poricanje odgovornosti: Informacije predstavljene u ovom članku o urođenim manama samo su za informiranje čitatelja. Nije namijenjen zamjeni za savjete zdravstvenog radnika.
