Amiloidoza sistemică: diagnostic, diagnostic diferențial, tratament. Cursul clinic și principalele sale manifestări
amiloidoza- o boală caracterizată printr-o tulburare metabolică, în urma căreia se formează o substanță nouă pentru organism (amiloid), care se depune în organe și le perturbă funcțiile.
Amiloidul este o glicoproteină complexă în care proteinele fibrilare și globulare sunt strâns asociate cu polizaharidele. Fibrila amiloidă este formată din proteine polipeptidice; Pe lângă proteina fibrilă, amiloidul conține și o altă proteină - așa-numita componentă P, care este aceeași în toate formele de amiloid. Se crede că componenta P este o proteină serică normală asociată cu fibrile de amiloid.
Amiloidoza poate apărea ca o complicație a oricărei boli sau se poate dezvolta ca un proces independent.
În prezent, în funcție de etiologie, se disting mai multe forme de amiloidoză, care au propria lor compoziție biochimică a fibrilelor de amiloid.
Amiloidoza primară (idiopatică) se dezvoltă fără motive vizibileși afectează diferite organe (inima, rinichii, intestinele, ficatul, sistemul nervos). Forma biochimică a amiloidozei primare este forma AL; precursorul unui astfel de amiloid este Ig și lanțurile uşoare de imunoglobuline. După structura amiloidului și natura leziunii organe interne Amiloidoza în mielomul multiplu, care este în prezent clasificată ca un grup separat, este aproape de doza primară (idiopatică) de amiloid.
Amiloidoza ereditară (genetică) se manifestă prin afectare predominantă a rinichilor, o combinație de afectare a rinichilor și sistem nervos. La noi, amiloidoza ereditară este de obicei asociată cu o boală periodică, care se transmite în mod autosomal dominant. În această boală, amiloidoza poate fi singura manifestare. Forma biochimică a amiloidozei ereditare este FA (precursorul amiloidului este prealbumina). În cazul bolilor periodice, forma biochimică este AA (precursorul este proteina SAA).
Amiloidoza dobândită (secundară) apare cel mai des și se dezvoltă cu poliartrită reumatoidă, spondilită anchilozantă, tuberculoză, supurație cronică - osteomielita, bronșiectazie, abces pulmonar cronic, mai rar - cu colită ulcerativă nespecifică, psoriazis, limfofiluloză, limfofiluloză etc. Forma biochimică a amiloidozei secundare este AA (precursorul său seric este proteina SAA, sintetizată de hepatocite).
Amiloidoza senilă este rezultatul unor tulburări involutive ale metabolismului proteinelor, găsite în creier, pancreas și inimă. Formula biochimică - AS (precursorul este prealbumina).
Amiloidoza locală se dezvoltă fără un motiv aparent, formula sa biochimică este AE (precursor necunoscut).
Patogeneza. Doar legăturile individuale ale patogenezei sunt bine cunoscute (Schema 22). Din diagramă rezultă că sub influența mutației genice, precum și influența factori externi imunitatea se modifică – scade
numărul de limfocite T. Acest lucru duce la o scădere a efectului lor de control asupra sistemului limfocitelor B. Ca rezultat, numărul de celule B care poartă imunoglobuline normale, iar numărul de celule B care sintetizează precursorii fibrilei amiloide crește. Amiloidoblastele produc proteine fibrilare în cantități crescute, ceea ce determină sinteza amiloidului în cantități mari.
Cu toate acestea, din cauza unui defect genetic la amiloidoclaste, care contribuie la scăderea activității lor enzimatice, nu are loc o resorbție suficientă a amiloidului. Ca rezultat, se observă o depunere crescută de amiloid în țesuturi și organe [Mukhin N.A., 1981].
În mielomul multiplu, amiloidoza se dezvoltă ca urmare a producției crescute de paraproteine de către celulele plasmatice, care merge spre construirea amiloidului. Compoziția amiloidului în diferite forme de amiloidoză este diferită, care este determinată de compoziția proteinei fibrilelor de amiloid.
Cu leziuni miocardice, nervi periferici(observat în principal în amiloidoza idiopatică) amiloidul se depune în jurul fibrelor de colagen țesut conjunctiv. În leziuni se observă depunerea de amiloid în jurul fibrelor reticulare
rinichi, intestine, ficat, glande suprarenale, pancreas (cu amiloidoză ereditară și secundară). Cu toate acestea, este posibilă o combinație de depunere de amiloid pericollagenos și perireticular, care oferă leziuni combinate ale diferitelor organe și sisteme.
Când amiloidul este depus în țesuturi, numărul de elemente funcționale, cardiomiocite, hepatocite, fibre nervoase și glomeruli renali scade, ceea ce duce ulterior la dezvoltarea insuficienței de organ.
Astfel, în inimă, amiloidul este depus sub endocard, în stroma și vasele miocardului, precum și de-a lungul venelor din epicard. În același timp, inima crește brusc în dimensiune, iar numărul de cardiomiocite scade rapid. Toate acestea duc la o scădere funcția contractilă miocard și insuficiență cardiacă, precum și tulburări de conducere și ritm cardiac. În creierul cu amiloidoză senilă, amiloidul se găsește în așa-numitele plăci senile ale cortexului, vaselor și membranelor. În piele, amiloidul se depune în papilele și pereții vaselor de sânge, ceea ce duce la atrofia severă a epidermei. În ficat, amiloidul se depune între reticuloendoteliocitele stelate ale vaselor sinusoidale, în pereții vaselor de sânge, ai canalelor și în țesutul conjunctiv al tractului portal. Pe măsură ce amiloidul se acumulează, celulele hepatice se atrofiază.
În rinichi, amiloidul se depune în membrana capilarelor glomerulare și a tubilor nefronici, în mezangiu, anse capilare și de-a lungul arteriolelor. Pe măsură ce amiloidul se acumulează, majoritatea nefronilor se atrofiază, mor sau sunt înlocuiți cu țesut conjunctiv - apare un rinichi cu riduri de amiloid. Acest proces poate fi reprezentat prin următoarea diagramă:
proteinurie -> sindrom nefrotic -> insuficienta renala.
În consecință, în tabloul clinic se disting trei etape: 1) inițială (proteinuric); 2) expandat (nefrotic); 3) terminal (azotemic).
Tabloul clinic. Manifestările amiloidozei sunt variate și sunt determinate de: 1) localizarea amiloidului într-un anumit organ; 2) severitatea depozitelor de amiloid în organ; 3) boala de bază împotriva căreia s-a dezvoltat amiloidul (în forma secundară de amiloidoză).
În timpul diagnosticului pot apărea dificultăți din cauza faptului că manifestările clinice ale bolii vor fi vizibile numai cu o anumită cantitate de amiloid depus. În acest sens, o perioadă „latentă” este inevitabilă din momentul depunerii de amiloid până la apariția simptomelor de disfuncție a unui organ sau sistem.
Tabloul clinic este deosebit de izbitor în cazurile de afectare a rinichilor, cel mai frecvent loc al depozitelor de amiloid.
La etapa I căutarea diagnosticului V stadiul inițial Aproape nu pot fi obținute informații care să indice afectarea rinichilor prin amiloidoză. Plângerile pacienților sunt legate de boala de bază (amiloidoză secundară).
Istoricul medical conține informații despre prezența unei anumite boli (tuberculoză pulmonară, osteomielita, artrită reumatoidă etc.), evoluția acesteia și terapia efectuată. Numai aceste informații nu permit diagnosticarea amiloidozei renale, dar alertează medicul cu privire la această posibilitate.
În stadiul avansat al amiloidozei, pacienții se plâng din cauza dezvoltării sindromului nefrotic, a scăderii cantității de urină, a edemului cu prevalență și severitate variate, precum și a plângerilor de slăbiciune, lipsă de apetit și scăderea performanței. Alături de ei, cu amiloidoza secundară există plângeri cu privire la manifestarea bolii de bază.
În stadiul terminal, plângerile sunt cauzate de dezvoltarea insuficienței renale cronice: pierderea poftei de mâncare, greață, vărsături (tulburări dispeptice), dureri de cap, tulburări de somn (tulburări ale sistemului nervos), mâncărime.
În stadiul II al căutării diagnostice, numai simptomele caracteristice bolii de bază (în amiloidoza secundară) pot fi detectate într-un stadiu incipient.
ÎN stagiu avansat detectează: 1) edem de diferite localizări și severitate; cu retenție semnificativă de lichide în organism, pot apărea hidrotorax, hidropericard și ascită tranzitorie; 2) hipertensiune arterială (apare la 12 - 20% dintre pacienții cu amiloidoză), dilatație și hipertrofie a ventriculului stâng; 3) mărirea ficatului și splinei datorită depunerii de amiloid în țesuturi (ficatul și splina sunt dense, nedureroase, cu marginea ascuțită); 4) simptome ale bolii de bază (cu amiloidoză secundară).
ÎN stadiu terminal simptomele sunt determinate de severitatea insuficienței renale: 1) sindrom distrofic (modificări ale pielii și mucoaselor); 2) sindrom sero-articular (osteoartropatie, gută secundară, pericardită uscată, pleurezie); 3) hipertensiune arterială.
În etapa III a căutării diagnostice pentru amiloidoză, se obțin cele mai semnificative informații pentru stabilirea unui diagnostic, care pot fi grupate în felul următor: 1) sindromul urinar; 2) tulburări ale metabolismului proteinelor și lipidelor; 3) detectarea depunerilor de mase de amiloid.
Sindromul urinar: 1) proteinurie - cel mai important simptom amiloidoza se dezvoltă în toate formele sale, dar cel mai adesea în amiloidoza secundară. Proteinuria este de obicei semnificativă; 2-20 g de proteine sunt eliberate pe zi, a cărei parte principală este albumina. Globulinele sunt eliberate în cantități mai mici, iar precursorul amiloid seric (proteina SAA) poate fi excretat prin urină. În stadiul terminal, proteinuria persistă. A- și mai ales γ-glicoproteinele pot fi găsite în urină.
În funcție de gradul de proteinurie, sunt detectate gipsuri hialine și, mai rar, granulare. Microhematuria sau leucocituria sunt rareori diagnosticate, dar severitatea acesteia nu corespunde gradului de proteinurie (cum se observă în cazul glomerulonefritei). Gradul de tulburări ale metabolismului lipidic în amiloidoză corespunde lipoiduriei cu prezența cristalelor birefringente în sedimentul urinar.
Tulburări ale metabolismului proteinelor și lipidelor: 1) hipoproteinemie în combinație cu hipoalbuminemia și hiper-<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
De mare importanță pentru diagnostic detectarea maselor de amiloid în organe și țesuturi:în ficat (50% din cazuri), splină (cu biopsie prin puncție), mucoasa gingiilor și rectului.
În stadiul inițial (etapa proteică), o biopsie a mucoasei gingiilor produce adesea un rezultat negativ, iar o biopsie a rectului - un rezultat pozitiv; în stadiul avansat (nefrotic), în primul caz rezultatele sunt pozitive la jumătate dintre pacienți, iar în al doilea - chiar mai des.
În cele din urmă, în insuficiența renală cronică, datele biopsiei din țesutul gingival sunt pozitive în mai mult de jumătate din cazuri și din mucoasa rectală în aproape toate cazurile. Prin urmare, ar trebui recomandată o biopsie a mucoasei gingiilor în caz de proces avansat și a rectului - în orice stadiu al amiloidozei.
Dacă se suspectează amiloidoza idiopatică (care afectează cel mai adesea inima, nervii periferici și mai rar rinichii), este indicat să se efectueze mai întâi o biopsie a mucoasei gingiilor, iar în cazul amiloidozei secundare (dobândite) și a formelor sale ereditare (procedând cu afectare primară a rinichilor), o biopsie a mucoasei rectului.
O serie de alte studii ajută: 1) la clarificarea diagnosticului bolii împotriva căreia s-a dezvoltat amiloidoza; 2) se evaluează starea funcțională a rinichilor (Rehberg, testul Zimnitsky, nivelul creatininei din sânge).
Curgere. Tabloul clinic al amiloidozei renale are caracteristici care o deosebesc de afectarea rinichilor de alte origini: 1) sindromul nefrotic se dezvoltă treptat și adesea după o etapă lungă de proteinurie, se caracterizează printr-un curs persistent, edemul este adesea rezistent la diferite diuretice. Cu CGN, sindromul nefrotic apare de obicei la debutul bolii și deseori recidivează în viitor; 2) hipertensiunea arterială se observă rar, chiar și în stadiul insuficienței renale cronice; 3) cu amiloidoza primară, insuficiența renală cronică este mai benignă în contrast cu amiloidoza secundară sau CGN (datorită severității mai mici a leziunii glomerulare în comparație cu formele secundare de amiloidoză); 4) cursul amiloidozei secundare depinde în mare măsură de boala de bază, cu exacerbări frecvente a căror progresie semnificativă a amiloidozei este posibilă.
Complicații. În cazul amiloidozei, în 2-5% din cazuri se dezvoltă următoarele:
1) tromboza venei renale (cu amiloidoza secundara), care se manifesta
prezinta hematurie si dureri in regiunea lombara, o crestere a proteinelor
ria și o scădere a diurezei;
2) infectie intercurente;
3) peritonita fibrinos-purulenta a carei aparitia este insotita de
este indicată de o creștere accentuată a ascitei.
Diagnosticare. Manifestările clinice ale amiloidozei sunt nespecifice. Fiecare dintre simptome (edem, proteinurie, hipertensiune arterială) poate apărea cu diferite boli de rinichi. Singura metodă de diagnosticare fiabilă a amiloidozei este o biopsie de organ (rinichi, ficat, mucoasă rectală sau gingie), dar nu este întotdeauna fezabilă. Prin urmare, în majoritatea cazurilor este necesar să se concentreze asupra manifestărilor clinice ale procesului patologic.
Prezența unei boli în care secundar
amiloidoza (semne clinice sau anamnestice).
Apariția și progresia proteinuriei sau apariția
sindrom nefrotic.
Nu există nicio boală care poate provoca amiloidoză,
cu toate acestea, proteinuria sau sindromul nefrotic este prezent.
Prezența insuficienței cardiace severe persistente, sindromul nu este
suficiență de absorbție, polineuropatie (dacă postnașterea
Apariția a trei sindroame este greu de explicat prin alte motive).
Prezența amiloidozei poate fi presupusă cu următoarele semne de laborator ale sindromului nefrotic (care, după cum se știe, se poate dezvolta în alte boli de rinichi):
a) disproteinemie severa + hipoalbuminemie + hiper-SSG- si hipo-
pergamaglobulinemie;
b) niveluri crescute de ag-glicoproteine, p-lipoproteine;
c) apariția în urină a a- și în special a γ-glicoproteinelor și a-lipopro-
Teidov.
În toate cazurile, probabilitatea dezvoltării amiloidozei crește odată cu detectarea hepato- și splenomegaliei, precum și a modificărilor cardiace caracteristice amiloidozei (în astfel de cazuri vorbim despre amiloidoza generalizată idiopatică).
În consecință, diagnosticul de amiloidoză renală poate fi pus cu suficientă încredere în stadiul avansat (nefrotic) sau terminal, în timp ce în stadiul inițial (proteinuric) acest lucru este mult mai dificil de realizat. In aceste cazuri, proteinuria tranzitorie sau permanenta trebuie diferentiata de glomerulonefrita (acuta, cronica). Trebuie luate în considerare următoarele:
1) progresie mai lentă a afectarii rinichilor cu amiloid
doza;
2) absența unei legături clare în amiloidoza cu răceli
niyami;
3) prezența constantă a microhematuriei în glomerulonefrită (cu
amiloidoză în 20% din cazuri).
Uneori, diagnosticul corect poate fi pus numai după o perioadă de observare pe termen lung a pacientului. Problema este rezolvată mult mai repede dacă este posibil să se efectueze o biopsie prin puncție a rinichiului.
Formularea unui diagnostic clinic detaliat amiloidoza are în vedere următoarele componente: 1) forma amiloidozei; 2) stadiul amiloidozei (proteinuric, nefrotic, terminal); 3) starea funcțională a rinichilor (absența sau prezența insuficienței renale, gradul de severitate a acesteia); 4) boala de bază (cu amiloidoză secundară); 5) starea altor organe (inima, ficat, sistem nervos etc.) cu amiloidoză idiopatică (primară).
Tratament. Problema tratării amiloidozei rămâne încă nerezolvată, deoarece motivele care duc la creșterea amiloidogenezei și la o resorbție insuficientă nu sunt clare. Cu toate acestea, este posibil să se efectueze o serie de măsuri terapeutice care îmbunătățesc starea pacientului. În prezent, tratamentul unui pacient cu amiloidoză se efectuează luând în considerare: 1) impactul asupra bolii de bază împotriva căreia s-a dezvoltat amiloidoza
(secundar); 2) influența asupra mecanismelor patogenezei; 3) impact asupra principalelor sindroame clinice.
Impact asupra bolii de bază împotriva căreia se dezvoltă
amiloidoza secundară este necesară datorită faptului că frecvente
tensiunea sau activitatea ridicată a procesului patologic duc la
progresia amiloidozei.
Acest impact este după cum urmează:
a) pentru infectii cronice (tuberculoza, sifilis) este necesar
terapie specifică pe termen lung;
b) pentru boli pulmonare cronice nespecifice - com
terapie complexă cu utilizarea antibioticelor, drenaj bronșic și
dacă este necesar, intervenție chirurgicală (de exemplu, pentru cronici
abces pulmonar logic);
c) cu boli sistemice ale țesutului conjunctiv, de exemplu cu
artrita reumatoida, este indicata terapia complexa, inclusiv
importanța medicamentelor de bază (D-penicilamină, săruri de aur, aminoacizi
medicamente noline).
Impactul asupra mecanismelor patogenezei implică reducerea
sinteza amiloidului:
a) aport zilnic de 80-120 g ficat crud timp de 6-12 luni
duce la o scădere a proteinuriei, o scădere a dimensiunii ficatului
nici splina;
b) medicamente aminochinoline (hingamină, sau delagil, conform
0,25 - 0,5 g pe zi timp de multe luni și chiar ani) se reduce
are loc o progresie a procesului. Aparent, aceste mijloace de influență
implicate în sinteza fibrilelor de amiloid. Tratamentul este eficient numai
în stadiile incipiente ale amiloidozei; când procesul este mult avansat
(sindrom nefrotic complet, insuficiență renală
ity) prescrierea acestor medicamente este inadecvată;
c) cu dezvoltarea amiloidozei din cauza bolii recurente periodice
Se recomanda colchicina;
d) in cazul amiloidozei primare se prescrie si melfalan, care suprima
împărtășind funcția unor clone de limfocite, în special syn
cele care sintetizează lanţurile uşoare ale imunoglobulinelor implicate în
formarea fibrilei de amiloid (aceasta se referă și la
amiloidoza care se dezvoltă în mielom).
Impactul asupra principalelor sindroame clinice implică
eliminarea edemului, a hipertensiunii arteriale, precum și a măsurilor care vizează
combaterea insuficienței renale în curs de dezvoltare:
a) cu dezvoltarea sindromului nefrotic și edem sever
este necesar un conținut suficient de proteine în alimente, reducerea
sare fiartă, precum și introducerea de sânge integral sau globule roșii
masă (mai ales în prezența anemiei), utilizare atentă
luarea de diuretice;
b) hipertensiunea arterială este mai puțin frecventă în amiloidoză,
cu toate acestea, atunci când ajunge la cifre mari, este necesar să se prescrie
introducerea diferitelor tipuri de medicamente antihipertensive;
c) dacă se dezvoltă insuficiență renală, tratamentul se efectuează conform planului general acceptat (restricționarea proteinelor în alimente, aport suficient de lichide, corectarea metabolismului mineral). Pentru insuficienta renala cauzata de amiloidoza, sunt posibile hemodializa si transplantul de rinichi.
Prognoza. Durata perioadei proteinurice este greu de determinat, cu toate acestea, după depistarea acesteia, edemul se dezvoltă de obicei după 3 ani, pe fondul căruia apare rapid insuficiența renală cronică. Toate acestea fac ca prognosticul să fie destul de grav.
Prevenirea. Pentru amiloidoza idiopatică și genetică, măsurile de prevenire primară sunt necunoscute. În cazul amiloidozei secundare, prevenirea constă în tratarea bolilor care duc la dezvoltarea amiloidozei.
RCHR (Centrul Republican pentru Dezvoltarea Sănătății al Ministerului Sănătății al Republicii Kazahstan)
Versiune: Protocoale clinice ale Ministerului Sănătății al Republicii Kazahstan - 2016
Amiloidoza (E85)
Nefrologie
Informații generale
Scurta descriere
Aprobat
Comisia mixtă pentru calitatea asistenței medicale
Ministerul Sănătății și Dezvoltării Sociale al Republicii Kazahstan
din 13 octombrie 2016
Protocolul nr. 13
amiloidoza- un grup de boli, al căror semn distinctiv este depunerea glicoproteinei fibrilare - amiloid - în țesuturi și organe.
Corelarea codurilor ICD-10 și ICD-9
| ICD-10 | ICD-9 | ||
| Cod | Nume | Cod | Nume |
| E85 | amiloidoza |
55.23 99.76 |
Biopsie renală închisă [percutanată] [punctură]. Hemodializa. Plasmafereza terapeutică Imunoadsorbția extracorporală |
| E85.0 | Amiloidoză familială ereditară fără neuropatie | ||
| E85.1 | Amiloidoza ereditară neuropatică | ||
| E85.2 | Amiloidoză ereditară, nespecificată | ||
| E85.3 | Amiloidoza sistemica secundara | ||
| E85.4 | Amiloidoză limitată | ||
| E85.8 | Alte forme de amiloidoză | ||
| E85.9 | Amiloidoza, nespecificata | ||
Data elaborării/revizuirii protocolului: 2016.
Utilizatori de protocol: Medici generalisti, terapeuți, hematologi, nefrologi.
Scala nivelului de evidență:
| A | O meta-analiză de înaltă calitate, o revizuire sistematică a RCT-urilor sau a RCT-urilor mari cu o probabilitate foarte scăzută (++) de părtinire, ale căror rezultate pot fi generalizate la o populație adecvată. |
| ÎN | Revizuirea sistematică de înaltă calitate (++) a studiilor de cohortă sau caz-control sau studii de cohortă sau caz-control de înaltă calitate (++) cu risc foarte scăzut de părtinire sau RCT cu risc scăzut (+) de părtinire, ale căror rezultate pot fi generalizate la o populație adecvată . |
| CU |
Studiu de cohortă sau caz-control sau studiu controlat fără randomizare cu risc scăzut de părtinire (+). Ale căror rezultate pot fi generalizate la populația relevantă sau RCT cu risc foarte scăzut sau scăzut de părtinire (++ sau +), ale căror rezultate nu pot fi generalizate direct la populația relevantă. |
| D | Serii de cazuri sau studiu necontrolat sau opinia unui expert. |
Clasificare
Tipuri de amiloid și forme corespunzătoare de amiloidoză:
|
Proteină amiloid |
Proteine precursoare | Forma clinică a amiloidozei |
| AA | proteina SAA | Amiloidoza secundară în bolile inflamatorii cronice, inclusiv boala periodică și sindromul Muckle-Wells |
| AL | λ, κ-lanțuri ușoare ale imunoglobulinelor | Amiloidoza în discraziile plasmatice - idiopatică, în mielomul multiplu și macroglobulinemia Waldenström |
| ATTR | Transtiretina | Forme familiale de amiloidoză polineuropatică, cardiopatică și alte amiloidoze, amiloidoză senilă sistemică |
| Ap2M | β2-microglobulina | Amiloidoza de dializă |
| AGel | Gelsolină | Polineuropatia amiloidă familială finlandeză |
| AApoAI | Apolipoproteina A-I | Polineuropatie amiloidă (tip III, conform lui van Allen, 1956) |
| AFib | Fibrinogen | Nefropatie amiloidă |
| Ap | proteina β | Boala Alzheimer, sindromul Down, hemoragii cerebrale ereditare cu amiloidoză, Olanda |
| APrP Scr | Proteine prionice | boala Creutzfeldt-Jakob, boala Gerstmann-Straussler-Scheinker |
| AANF | Factorul natriuretic atrial | Amiloidoza atrială izolată |
| AIAPP | amilină | Amiloidoza izolata in insulele Langerhans cu diabetul zaharat Tipul II, Insulinom |
| ACal | Procalcitonina | Pentru cancerul medular glanda tiroida |
| ACys | Cistatina C | Hemoragii cerebrale ereditare cu amiloidoză, Islanda |
Clasificarea clinică a amiloidozei
amiloidoza primara:
· care apar fără un motiv aparent;
asociat cu mielom multiplu;
amiloidoza secundara:
· pentru infectii cronice;
· pentru artrita reumatoidă și alte boli ale țesutului conjunctiv;
· pentru boli oncologice;
amiloidoza familială (ereditară):
· cu boli periodice;
· Varianta portugheză și alte forme de amiloidoză familială;
amiloidoza senilă
amiloidoza locala
amiloidoza ereditara:
neuropatic
· cu afectarea extremităților inferioare: portugheză, japoneză, suedeză și alte tipuri;
· cu afectare a extremităților superioare: tipuri Elveția-Indiana, Germania-Maryland;
nefropat:
· boli periodice;
· febră și dureri abdominale la suedezi și sicilieni;
combinație de erupție cutanată, surditate și leziuni renale;
· afectarea rinichilor in combinatie cu hipertensiunea arteriala;
cardiomiopatice:
daneză - insuficiență cardiacă progresivă;
· Mexican-American - sindromul sinusului bolnav, stop atrial;
amestecat:
finlandeză - distrofie corneană și afectarea nervilor cranieni;
· accidente vasculare cerebrale.
Stadiile clinice ale amiloidozei renale
| Etapă | Manifestare clinică |
| 1 | Stadiul preclinic sau latent (asimptomatic) - amiloidul este prezent în zona intermediară și edem și focare de scleroză se dezvoltă de-a lungul vaselor drepte ale piramidelor. Etapa durează 3-5 sau mai mulți ani. În această perioadă, cu amiloidoza reactivă, predomină manifestările clinice ale bolii de bază (de exemplu, proces purulent în plămâni, tuberculoză, artrită reumatoidă etc.). |
| 2 | Stadiul proteinuric (albuminuric) - amiloidul apare în primul rând în mezangiu, în ansele capilare, în piramidele și cortexul glomerulilor, în vase. Se dezvoltă scleroza și atrofia nefronului, hiperemia și limfostaza. Mugurii sunt măriți și denși, de culoare gri-roz mat. Proteinuria la început este moderat exprimată, poate fi chiar tranzitorie pentru o anumită perioadă, scade și crește, dar apoi devine persistentă (stadiul proteinuriei intermitente). Unii cercetători disting două perioade în acest stadiu: proteinuria selectivă și neselectivă. Durata etapei este de la 10 la 13 ani. |
| 3 | Stadiul nefrotic (edematos, edematos-hipotonic) - nefroza amiloid-lipoid - amiloid în toate părțile nefronului. Există scleroză și amiloidoză medulară, dar stratul cortical este fără modificări sclerotice pronunțate. Durata etapei este de până la 6 ani. Atât în stadiul proteinuric, cât și în cel nefrotic, rinichii sunt măriți și denși (rinichi sebaceu mare). Clinic, această etapă se manifestă prin sindromul nefrotic clasic cu toate semnele sale: cu dezvoltarea proteinuriei masive (cu pierderi de proteine în urină de peste 3-5 grame pe zi), hipoproteinemie cu hipoalbuminemie, hipercolesterolemie, lipidurie cu edem la gradul de anasarca. În sedimentul urinar se găsesc gipsuri hialine, iar pe măsură ce proteinuria crește, se găsesc gipsuri granulare. Sunt posibile micro- și macrohematurie și leucociturie fără semne de pielonefrită. |
| 4 | Stadiul uremic (terminal, azotemic) - rinichi amiloid ridat - redus ca dimensiune, dens, cu cicatrici. Insuficiența renală cronică diferă puțin de cea a altor boli de rinichi. Se crede că, spre deosebire de glomerulonefrită, în care debutul insuficienței renale cronice, care apare cu poliurie, poate duce la o convergență cel puțin parțială a edemului, cu amiloidoză, azotemia se dezvoltă pe fondul tensiunii arteriale scăzute și al sindromului nefrotic. |
Diagnostice (ambulatoriu)
DIAGNOSTICUL AMBULATORULUI
Criterii de diagnostic
Reclamații:
slăbiciune, oboseală crescută;
· durere de cap;
· umflarea picioarelor, brațelor și feței;
· tensiune arterială crescută;
Greață, diaree (diaree);
· durere în zona inimii;
· dureri musculare.
Anamneză:
· pierdere în greutate;
· prezența gamapatiei monoclonale de origine necunoscută;
· boli inflamatorii cronice (purulente);
· infectii cronice;
· ereditatea.
Examinare fizică
Inspecție generală:
· purpură periorbitală (observată în 15% din cazuri);
Macroglosia este caracteristică amiloidozei primare (AL);
· dificultăți de respirație în timpul efortului fizic (observată la aproximativ 40% dintre pacienți);
· semnul umărului (infiltrarea periarticulară de amiloid duce la falsă hipertrofie și la creșterea volumului mușchilor centurii scapulare și coapsei).
Auscultatie:
Poate exista o tulburare de ritm cardiac.
Palpare:
· edem al extremităților inferioare, din cauza hipoalbuminemiei și sindromului nefrotic, precum și din cauza stagnării circulației sistemice din cauza cardiomiopatiei restrictive (observată în 50% din cazuri);
Ficat și splina mărite;
· parestezii (observate la aproximativ 15% dintre pacienți);
· durere spasmodică în tractul gastrointestinal;
· poate exista o marire a glandelor salivare submandibulare.
Cercetare de laborator:
· hemoleucograma completă - anemie, leucocitoză, VSH crescut;
· analiza generala a urinei - proteinurie, microhematurie, leucociturie aseptica;
· test biochimic de sânge (proteine totale, albumină, Na, Ca, colesterol, zahăr din serul sanguin) - hipoproteinemie (datorită hipoalbuminemiei), hiperglobulinemie, hiponatremie, hipoprotrombinemie, hipocalcemie, hipercolesterolemie.
· Ecografia organelor cavitate abdominală iar rinichii - sunt vizualizati rinichi mariti, compactati (rinichi grasi mari).

Diagnosticare (spital)
DIAGNOSTICĂ LA NIVEL DE PACIENȚI STATINAT
Criterii de diagnostic la nivel de spital
Reclamații și anamneză: vezi nivelul ambulatoriului.
Examinare fizică: vezi nivelul ambulatoriului.
Cercetare de laborator:
| Test de diagnosticare | Rezultat |
|
Imunofixarea serică Testul este pozitiv la 60% dintre pacienții cu amiloidoză cu lanț ușor (AL) de imunoglobuline (6). |
|
|
Imunofixarea urinei Testul este pozitiv la 80% dintre pacienții cu amiloidoză AL (6). Detectarea proteinei lanțului ușor în urină sugerează prezența mielomului multiplu și a amiloidozei. |
Prezența proteinei monoclonale |
|
Testarea imunoglobulinelor cu lanț ușor liber în ser Acest test relativ nou cu foarte sensibilitate crescută(>95%) pentru diagnosticul amiloidozei AL (10). Antiserurile disponibile comercial pentru lanțurile ușoare de imunoglobuline, AA și transtiretina sunt în general utilizate, dar pot lipsi suficientă specificitate și sensibilitate. În multe cazuri, spectroscopia de masă și microscopia imuno-electronică sunt necesare pentru a determina tipul de amiloid subiacent. |
Raport kappa lambda anormal |
|
Biopsie măduvă osoasă
O biopsie de măduvă osoasă se efectuează la toți pacienții cu suspiciune de amiloidoză cu lanț ușor și este sursa grozavațesut pentru a diagnostica orice pacient cu suspiciune de amiloidoză. |
Prezența celulelor plasmatice clonale |
Studii instrumentale:
· Ecografia organelor abdominale si a rinichilor - se vizualizeaza rinichi mariti, compactati (rinichi grasi mari).
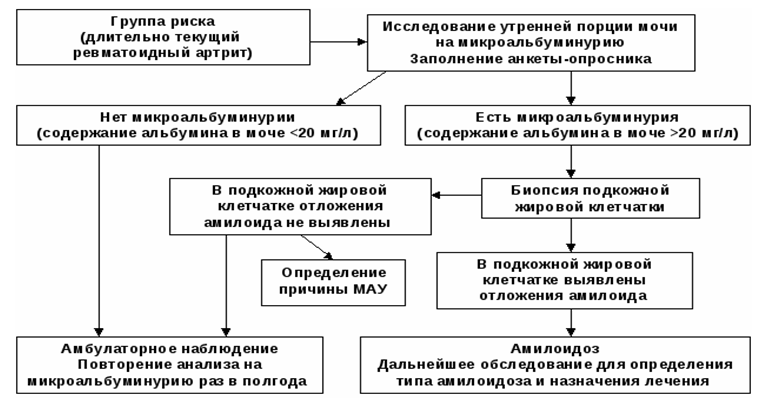
Algoritm de diagnostic pentru amiloidoza renală
Lista principalelor măsuri de diagnostic:
· analize generale de sânge;
· analiza generală a urinei;
· test biochimic de sânge (proteine totale, albumină, Na, Ca, colesterol, zahăr din sânge);
Imunofixarea serică;
· imunofixarea urinei;
· studiul imunoglobulinelor cu lanț ușor liber din ser;
Biopsie de măduvă osoasă.
· Ecografia organelor abdominale si rinichilor.
Lista măsurilor suplimentare de diagnosticare
Cercetare de laborator:
| Test de diagnosticare | Rezultat |
|
Biopsie tisulară: Pentru a diagnostica amiloidoza, depozitele de țesut din materialul de biopsie trebuie colorate pozitiv cu roșu Congo (11). Birefringența verde strălucitor poate fi văzută atunci când materialul Congo este colorat în roșu sub lumină polarizată. Materialul de biopsie poate fi obținut din mucoasa buzelor, piele, gingii, grăsime subcutanată, măduvă osoasă, nervi, rect, rinichi, ficat sau inimă. Depozitele sunt intotdeauna localizate extracelular si sunt amorfe. |
pozitiv - birefringență verde atunci când este colorată cu roșu Congo |
|
Studii imunohistologice ale depozitelor de amiloid: Ele permit recunoașterea diferitelor forme de amiloidoză sistemică. |
antiser pentru lanțul ușor de imunoglobuline, AA și transtiretină |
|
Spectroscopie de masă: Oferă analiza compoziției proteinei amiloide. În prezent, standardul de aur pentru diagnosticarea tipului de amiloidoză. |
confirmă tipul de proteină |
|
Microscopia imunoelectronică: Toate formele de amiloid microscop electronic fibros, dur și neramificat. |
amiloizii au aspect fibrilar și sunt rigizi și neramificați. |
|
Testare genetică: Pentru a exclude amiloidoza ereditară în cazul rezultatelor testelor îndoielnice pentru detectarea proteinei imunoglobulinei monoclonale/lanțului ușor de amiloid liber, testarea genetică este obligatorie. Genele pot fi examinate prin secvențiere directă și includ următoarele gene: TTR, fibrinogen, apolipoproteina A1, lizozimă, MEFV (febră mediteraneană) și receptorul 1 al factorului de necroză tumorală (TNFR1 sau TNFRSF1A). MEFV și TNFRSF1A sunt sindroame moștenite febră periodică(adică, cauze potențiale ale amiloidozei AA) și nu sunt amiloidoze ereditare în sine. |
pozitiv |
|
Scanarea scintigrafică cu amiloid P seric (SAP): În ultimii ani, scintigrafia cu componenta P seric marcat cu iod (SAP) a fost utilizată în practica clinică pentru a evalua distribuția amiloidului în organism. |
captarea la locurile de depunere de amiloid |
|
Analiza generala sânge: Anemia se observă în principal la pacienții cu insuficiență renală sau sângerare din tractul gastrointestinal. Trombocitemia este asociată cu afectarea ficatului și hipersplenism. |
de obicei normal |
|
Test biochimic de sânge (teste hepatice și renale, indicatori ai stării metabolice): Amiloidoza hepatică se caracterizează prin niveluri crescute fosfataza alcalină. Majoritatea pacienților cu amiloidoză renală în stadiu incipient mențin clearance-ul creatininei, dar pot prezenta grade semnificative de hipoalbuminemie din cauza pierderii de proteine în urină (sindrom nefrotic). |
Niveluri scăzute de albumină; fosfatazei alcaline crescute |
|
proteinurie zilnică (colectare de urină 24 de ore): Excreția de albumină > 1 g/zi la pacienții cu amiloidoză indică leziuni renale (amiloidoză renală). Când nivelurile de proteinurie sunt > 3 g/zi, se dezvoltă sindromul nefrotic. |
creșterea proteinelor în urină |
|
Nivelul troponinei serice: Test sensibil pentru a detecta leziuni miocardice. Pacienții cu niveluri de troponine detectabile au un prognostic mai rău decât cei fără acestea (12). |
elevat |
|
Peptida natriuretică de tip B: Test de diagnostic sensibil pentru prezența întinderii miocardice și a ICC. S-a dovedit a fi important valoare de prognosticîn stabilirea amiloidozei cardiace (13). La niveluri de peptide natriuretice de tip B >300 ng/L (>300 pg/ml) sugerează implicarea amiloidului miocardic (10). Pacienții cu<170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с >170 ng/l (>170 pg/ml). |
elevat |
|
microglobuline beta-2: Este un predictor al supraviețuirii la pacienții cu amiloidoză. Nivelurile de beta-2-microglobuline >2,7 mg/L au un prognostic prost (14). |
elevat |
Studii instrumentale:
|
ECG: Ar trebui efectuat la toți pacienții ca parte a evaluării implicării cardiace. |
tulburare de conducere cardiacă |
|
Ecocardiograma (EchoCG): Semnele clinice de insuficiență cardiacă la pacienții cu amiloidoză cardiacă apar în 22% până la 34% (7). Ecocardiografia relevă o incidență mare a depunerilor de amiloid la pacienții cu simptome minime (aproximativ 50% din cazurile de AL au afectare cardiacă). ÎN ultima etapă are loc o scădere a fracției de ejecție. |
disfuncție diastolică, îngroșarea septului interventricular, scăderea fracției de ejecție |
|
Echo Dopplerografie cu tensiune: Un indicator al gradului de infiltrație de amiloid în miocard. Are o sensibilitate ridicată pentru detectarea anomaliilor atunci când nu există hipertensiune sau boală valvulară. Întinderea miocardică este definită ca modificarea procentuală a lungimii fibrei miocardice pe unitatea de lungime, iar rata depinde de durata întinderii (15-16). |
Contractia longitudinala redusa si intinderea miocardului; limitarea umplerii camerei ventriculare |
|
RMN al inimii: Relaxometria prin rezonanță magnetică îmbunătățește fiabilitatea diagnosticului RMN și ajută la distingerea amiloidozei cardiace de cardiomiopatia hipertrofică. |
a crescut semnificativ timpii de relaxare T1 și T2 în comparație cu controalele legate de vârstă |
Diagnostic diferentiat
Diagnostic diferentiat amiloidoza renala
| Stat | Semne/simptome diferențiabile | Teste diferențiabile |
| Cardiomiopatie hipertropica | Din punct de vedere clinic, este dificil să distingem HCM de amiloidoza cardiacă. |
· Ecocardiografia este criteriu de diagnostic pentru GCM, unde este detectată hipertrofia asimetrică a septului interventricular; · ecoul Doppler de tensiune utilizat pentru a exclude semnele de amiloidoză nu indică modificările tipice restrictive ale umplerii detectate în amiloidoză; · RMN-ul permite distingerea a 2 sindroame |
| Glomerulopatia membranoasă | Manifestări clinic identice la pacienții cu sindrom nefrotic. | · biopsia renală nu este colorată cu roșu Congo. |
| Gammopatie monoclonală de origine necunoscută (MGUS) neuropatie asociată | Pacienții nu prezintă proteinurie semnificativă, hepatomegalie sau cardiomiopatie. | · biopsia nervului sural nu se colorează cu roșu Congo. |
| Mielom multiplu | Dureri osoase, simptome de anemie și insuficiență renală. |
· obișnuit raze X prezintă leziuni osoase litice, fracturi de compresie, osteoporoza difuza; · Hb scăzut; · insuficiență renală. |
| Sindrom nefrotic | proteinurie zilnică mai mare de 3,5 g/zi, edem, hipoalbuminemie, dislipidemie | · vezi KP „Sindromul nefrotic” |
Turism medical
Obțineți tratament în Coreea, Israel, Germania, SUA
Turism medical
Obțineți sfaturi despre turismul medical
Tratament
Atenţie!
- Prin auto-medicație, puteți provoca vătămări ireparabile sănătății dumneavoastră.
- Informațiile postate pe site-ul web MedElement nu pot și nu trebuie să înlocuiască consultație față în față doctor Asigurați-vă că contactați institutii medicale dacă aveți boli sau simptome care vă deranjează.
- Alegerea medicamentelor și doza acestora trebuie discutate cu un specialist. Doar un medic poate prescrie medicamentul potrivit și doza acestuia, ținând cont de boala și starea corpului pacientului.
- Site-ul web MedElement este doar o resursă de informare și referință. Informațiile postate pe acest site nu trebuie folosite pentru a modifica în mod neautorizat comenzile medicului.
- Editorii MedElement nu sunt responsabili pentru nicio vătămare corporală sau daune materiale rezultate din utilizarea acestui site.
- general, boala sistemica organism, în care depunerea unei glicoproteine specifice (amiloid) are loc în organe și țesuturi cu funcția afectată a acesteia din urmă. Amiloidoza poate afecta rinichii (sindrom nefrotic, sindrom de edem), inima (insuficiență cardiacă, aritmii), tractul gastrointestinal, sistemul musculo-scheletic și pielea. Posibila dezvoltare a poliserozitei, sindromul hemoragic, probleme mentale. Diagnosticul de încredere al amiloidozei este facilitat de detectarea amiloidului în probele de biopsie ale țesuturilor afectate. Pentru a trata amiloidoza, se efectuează terapie imunosupresoare și simptomatică; dupa indicatii - dializa peritoneala, transplant de rinichi si ficat.
ICD-10
E85

Informații generale
Amiloidoza este o boală din grupul disproteinozelor sistemice care apare odată cu formarea și acumularea în țesuturi a unui compus complex proteic-polizaharidic - amiloid. Prevalența amiloidozei în lume este în mare măsură determinată geografic: de exemplu, boala periodică este mai frecventă în țările din bazinul mediteranean; polineuropatia amiloidă - în Japonia, Italia, Suedia, Portugalia etc. Frecvența medie amiloidoza în populație este de 1 caz la 50 mii populație. Boala se dezvoltă de obicei la persoanele cu vârsta peste 50-60 de ani. Dat fiind faptul că amiloidoza afectează aproape toate sistemele de organe, boala este studiată de diverse discipline medicale: reumatologie, urologie, cardiologie, gastroenterologie, neurologie etc.
Cauzele amiloidozei
Etiologia amiloidozei primare nu a fost studiată pe deplin. Cu toate acestea, se știe că amiloidoza secundară este de obicei asociată cu boli infecțioase cronice (tuberculoză, sifilis, actinomicoză) și purulent-inflamatorii (osteomielita, bronșiectazie, endocardită bacteriană etc.), mai rar - procesele tumorale(limfogranulomatoză, leucemie, cancer de organe viscerale). Amiloidoza reactivă se poate dezvolta la pacienții cu ateroscleroză, psoriazis, patologie reumatică (artrita reumatoidă, spondilită anchilozantă), inflamație cronică(colita ulcerativa nespecifica, boala Crohn), leziuni multisistemice (boala Whipple, sarcoidoza). Printre factorii care contribuie la dezvoltarea amiloidozei, hiperglobulinemia, funcționarea afectată a imunității celulare, predispoziția genetică etc. sunt de o importanță capitală.
Patogeneza
Printre numeroasele versiuni ale amiloidogenezei cel mai mare număr Teoriile disproteinozei, geneza celulară locală, teoriile imunologice și mutaționale au susținători. Teoria genezei celulare locale ia în considerare doar procesele care au loc la nivel celular (formarea de precursori de amiloid fibrilar de către sistemul macrofag), în timp ce formarea și acumularea de amiloid are loc în afara celulei. Prin urmare, teoria genezei celulelor locale nu poate fi considerată exhaustivă.
Conform teoriei disproteinozei, amiloidul este un produs al metabolismului anormal al proteinelor. Principalele legături în patogeneza amiloidozei - disproteinemia și hiperfibrinogenemia - contribuie la acumularea de fracții grosiere de proteine și paraproteine în plasmă. Teoria imunologică a originii amiloidozei asociază formarea amiloidului cu reacția antigen-anticorp, în care antigenele sunt proteine străine sau produși de degradare ai propriilor țesuturi. În acest caz, depunerea de amiloid are loc predominant în zonele de formare a anticorpilor și excesul de antigene. Cea mai universală este teoria mutației amiloidozei, care ia în considerare o mare varietate de factori mutageni care pot provoca sinteza anormală a proteinelor.
Amiloidul este o glicoproteină complexă formată din proteine fibrilare și globulare strâns asociate cu polizaharide. Depozitele de amiloid se acumulează în intima și adventiția vaselor de sânge, stroma organelor parenchimatoase, structurile glandulare etc. În cazul depozitelor de amiloid minore, modificările sunt detectate doar la nivel microscopic și nu duc la tulburări funcționale. Acumularea severă de amiloid este însoțită de modificări macroscopice ale organului afectat (volum crescut, aspect gras sau ceros). Ca urmare a amiloidozei, se dezvoltă scleroza stromală și atrofia parenchimului de organ și eșecul lor funcțional semnificativ clinic.
Clasificare
După cauze, se disting amiloidoza primară (idiopatică), secundară (reactivă, dobândită), ereditară (familială, genetică) și senilă. Există diferite forme de amiloidoză ereditară: febră mediteraneană sau boli periodice (crise de febră, dureri abdominale, constipație, diaree, pleurezie, artrită, erupții cutanate), amiloidoză neuropatică portugheză (polineuropatie periferică, impotență, tulburări de conducere cardiacă), tip finlandez ( atrofia corneei, neuropatie craniană), varianta daneză (amiloidoză cardiopatică) și multe altele. etc.
În funcție de afectarea predominantă a organelor și sistemelor, nefropatic (amiloidoza rinichilor), cardiopatic (amiloidoza inimii), neuropatic (amiloidoza sistemului nervos), hepapatic (amiloidoza ficatului), epinefropatic (amiloidoza glandelor suprarenale). ), se disting amiloidoza ARUD, amiloidoza pielii și un tip mixt de boală. În plus, în practica internațională se obișnuiește să se facă distincția între amiloidoza locală și cea generalizată (sistemică). La forme localizate, de obicei în curs de dezvoltare la indivizi in varsta, includ amiloidoza în boala Alzheimer, diabetul zaharat de tip 2, tumorile endocrine, tumorile de piele, Vezica urinara etc În funcţie de compoziție biochimică Printre formele sistemice de amiloidoză, se disting următoarele tipuri de fibrile amiloide:
- AL- în compoziţia fibrilelor lanţuri uşoare Ig (pentru boala Waldenström, mielom multiplu, limfoame maligne);
- A.A.– fibrilele conțin α-globulină serică de fază acută, asemănătoare prin caracteristici cu proteina C reactivă (pentru afecțiuni tumorale și reumatismale, boli periodice etc.);
- Ap2M- contine fibrile de β2-microglobulina (pentru insuficienta renala cronica la pacientii in hemodializa);
- ATTR– fibrilele conţin proteina de transport transtiretină (în formele familiale ereditare şi senile de amiloidoză).
Simptomele amiloidozei
Manifestările clinice ale amiloidozei sunt diverse și depind de severitatea și localizarea depozitelor de amiloid, de compoziția biochimică a amiloidului, de durata bolii și de gradul de disfuncție de organ. În stadiul latent al amiloidozei, când depozitele de amiloid pot fi detectate doar microscopic, nu există simptome. Pe măsură ce insuficiența funcțională a unui anumit organ se dezvoltă și progresează, semnele clinice ale bolii cresc.
Cu amiloidoza renală, stadiul pe termen lung al proteinuriei moderate este înlocuit cu dezvoltarea sindromului nefrotic. Trecerea la stadiul avansat poate fi asociată cu o infecție intercurrentă, vaccinare, hipotermie sau exacerbare a bolii de bază. Umflarea crește treptat (mai întâi la nivelul picioarelor și apoi în tot corpul), se dezvoltă hipertensiunea arterială nefrogenă și insuficiența renală. Poate apărea tromboză venoasă renală. Pierderea masivă de proteine este însoțită de hipoproteinemie, hiperfibrinogenemie, hiperlipidemie și azotemie. Micro-, uneori macro-hematurie și leucociturie sunt detectate în urină. În general, în timpul amiloidozei renale, se disting un stadiu precoce needematos, un stadiu edemat și un stadiu uremic (cahectic).
Amiloidoza cardiacă apare ca o cardiomiopatie restrictivă cu semne clinice tipice - cardiomegalie, aritmie, insuficiență cardiacă progresivă. Pacienții se plâng de dificultăți de respirație, umflături și slăbiciune care apare la un efort fizic minor. Mai rar, cu amiloidoza cardiacă, se dezvoltă poliserozită (ascita, pleurezie exudativă și pericardită).
Afectarea tractului gastro-intestinal în amiloidoză se caracterizează prin infiltrarea amiloidă a limbii (macroglassie), esofagului (rigiditate și afectarea peristaltismului), stomacului (arsuri la stomac, greață), intestine (constipație, diaree, sindrom de malabsorbție, obstrucție intestinală). Sângerările gastrointestinale pot apărea la diferite niveluri. Odată cu infiltrarea amiloidă a ficatului, se dezvoltă hepatomegalie, colestază și hipertensiune portală. Leziunile pancreasului din cauza amiloidozei sunt de obicei deghizate ca pancreatită cronică.
Amiloidoza cutanată apare cu apariția mai multor plăci de ceară (papule, noduli) la nivelul feței, gâtului și pliurilor naturale ale pielii. Prin semne externe, leziunile cutanate pot semăna cu sclerodermia, neurodermatita sau lichenul plan. Pentru leziunile amiloide ale sistemului musculo-scheletic, dezvoltarea poliartritei simetrice, carpiene sindromul de tunel, periartrita glenohumerala, miopatie. Forme individuale amiloidoza care implică sistemul nervos poate fi însoțită de polineuropatie, paralizie a membrelor inferioare, dureri de cap, amețeli, hipotensiune ortostatică, transpirație, demență etc.
Diagnosticare
Diferiți clinicieni pot întâlni manifestările clinice ale amiloidozei: reumatologi, urologi, cardiologi, gastroenterologi, neurologi, dermatologi, terapeuți etc. De importanță primordială pentru setarea corectă Diagnosticul se bazează pe o evaluare cuprinzătoare a semnelor clinice și anamnestice și pe o examinare cuprinzătoare de laborator și instrumentală.
Amiloidoza este o boală sistemică; afectează metabolismul proteinelor, sistemul imunitar nu mai funcționează. În acest sens, se formează amiloid - un complex proteină-zaharidă care se depune în toate țesuturile organelor umane.
În timp, amiloidul afectează tot mai mult organele, înlocuind celulele normale. Ca urmare, organul își pierde funcționalitatea și modificări ireversibile. Dacă boala nu este tratată pentru o lungă perioadă de timp, funcțiile mai multor organe sunt afectate, ceea ce duce la moarte.
Conform cercetărilor OMS, amiloidoza este diagnosticată la aproximativ 1% dintre locuitorii lumii. Amiloidoza secundară este cea mai frecventă. Amiloidoza genetică este mai des diagnosticată la persoanele care aparțin naționalității evreiești, armeane, precum și la rezidenții țărilor mediteraneene.
Rata de incidență în rândul bărbaților este de două ori mai mare decât în rândul femeilor. Dintre toate formele de amiloidoză, se diagnostichează amiloidoza nefropatică (lezarea rinichilor) și generalizată (lezarea tuturor țesuturilor și organelor).
Tipuri de amiloidoză, cauze de dezvoltare
În funcție de cauza amiloidozei, există tipuri de boli, care se pot dezvolta independent sau din cauza patologiilor în alte sisteme și organe. Întâlni următoarele tipuri amiloidoza: senila, cu tumori, amiloidoza primara sau idiopatica, ereditara, secundara sau reactiva, precum si la pacientii supusi hemodializa. În funcție de tip, dezvoltarea amiloidozei are loc diferit, simptomele și prognosticul variază. Mai jos vom discuta în detaliu tipurile și etapele amiloidozei.
primar (idiopatic)
Amiloidoza primară în cele mai multe cazuri începe fără motiv. În această formă a bolii, amiloidul este depus în țesuturi și organe și se observă o mutație a celulelor sistemului imunitar. AL-amiloidul format în acest proces se acumulează în mușchi, piele, sistemul cardiovascular și nervi. De asemenea, amiloidul AL se formează pe fondul mielomului tumoral, când celulele plasmatice încep să secrete globuline în volume mari. După legarea de nucleoproteinele plasmatice, globulinele anormale sunt transformate în amiloid.
Secundar (reactiv)
Amiloidoza secundară se dezvoltă pe fondul proceselor inflamatorii progresive în timp. În acest caz, amiloidul AA se formează ca o complicație a altor boli. Motivele pentru care apare amiloidoza secundară sunt:
- Infecții cronice - lepră, malarie, tuberculoză, sifilis, pielonefrită, bronșiectazie.
- Boli cronice purulente - supurația rănilor pe o perioadă lungă de timp, osteomielita.
- Tumori – leucemie, limfogranulomatoză etc.
- Prezența colitei ulcerative nespecifice (inflamația intestinului gros).
- Boală reumatologică – spondilită anchilozantă, artrită reumatoidă etc.
Amiloidoza secundară poate afecta orice organ sau țesut din organism. Manifestarea bolii nu este imediat vizibilă. La ani de la debutul bolii de bază, se poate observa o disfuncție a organului unde se depune cel mai mult amiloidul. Ficatul, rinichii, splina și ganglionii limfatici sunt cel mai adesea afectați de această tulburare. În timp, alte organe sunt afectate, ceea ce duce la insuficiență de organe multiple și la moarte.
Amiloidoza ereditară
Forma ereditară a amiloidozei este cauzată de prezența genelor mutante în celulele sistemului imunitar. Aceste mutații genetice sunt transmise de-a lungul generațiilor, ducând la formarea de amiloidoblaste. Forma ereditară afectează oamenii dintr-o anumită zonă sau aparținând unei anumite grup etnic. Amiloidoza ereditară este împărțită în tipuri:
- Cardiopatică. Este diagnosticată în principal la rezidenții din Danemarca. Tabloul clinic al bolii seamănă cu amiloidoza primară de tip generalizat.
- neuropatic. Caracterizat prin înfrângere țesut nervos. În funcție de localizarea leziunii, există amiloidoză portugheză (nervii picioarelor), americană (nervii brațelor), finlandeză (sistem nervos, cornee, rinichi).
- Nefropat familial. Un alt nume este amiloidoza engleză (boala Muckle și Wells). Tabloul clinic este urticarie, crize de febră, tulburări de auz.
- Periodic (febra mediteraneană familială). Boala este mai frecventă printre evrei, arabi și armeni. Manifestări – temperatură peste 39ºС, durere în cap și mușchi, transpirație abundentă. Se observă inflamația membranelor plămânilor, a organelor peritoneale și a organelor sinoviale. Anomaliile mentale sunt frecvente.
Amiloidoza senilă
La persoanele cu vârsta peste 80 de ani, amiloidul se depune local în diferite țesuturi și organe. Boala este asociată cu alte boli legate de vârstă. Există două tipuri de amiloidoză senilă:
- Cerebral sau cerebral. Se dezvoltă pe fondul bolii Alzheimer. Amyloid Ab este depus în țesutul cerebral.
- Cordial. Poate afecta ventriculii inimii (când amiloidul este format dintr-o proteină mutantă din sânge transtiretină) și atriile (când amiloidul este format din peptida natriuretică secretată de celulele cardiace). În ambele cazuri, amiloizii se găsesc în țesuturile plămânilor, pancreasului și splinei.
Pentru tumori
Unele tipuri de tumori afectează transformarea malignă a celulelor organului bolnav, care, ca urmare, produc proteine fibrilare. În acest caz, amiloidoza se dezvoltă local în țesutul organului afectat de tumoră. Motive care provoacă amiloidoza în tumori:
- Tumora medulară a glandei tiroide. Cancerul se dezvoltă din celulele C ale glandei tiroide, care instare buna responsabil de producerea calcitocinei. Când sinteza calcitocinei este întreruptă, fragmentele sale devin parte din AE amiloid.
- Cancerul insulelor tiroidiene. Insulele sunt grupuri de celule responsabile de producerea de hormoni - glucagon, insulină, somatostatina etc. Degenerarea malignă a celulelor determină eliberarea proteinei fibrilare, care ulterior degenerează în amiloid.
Amiloidoza în timpul hemodializei
 Hemodializa este un tratament care salvează vieți pentru pacienții ai căror rinichi nu sunt capabili să elimine toxinele din sânge. produse secundare metabolism. Hemodializa este prescrisă celor diagnosticați cu insuficiență renală (acută, cronică).
Hemodializa este un tratament care salvează vieți pentru pacienții ai căror rinichi nu sunt capabili să elimine toxinele din sânge. produse secundare metabolism. Hemodializa este prescrisă celor diagnosticați cu insuficiență renală (acută, cronică).
Esența procedurii este trecerea sângelui printr-o mașină care elimină substanțele nocive din acesta, returnând sângele purificat în corpul pacientului.
În timpul dializei, B2-microglobulina nu poate fi îndepărtată din organism și dacă pacientul perioadă lungă de timp forțat să facă hemodializă, proteinele se acumulează în organism în cantități excesive. Se leagă de nucleoproteinele plasmatice și se instalează diferite organe, devine baza amiloidului.
Simptomele amiloidozei
Având în vedere că boala se poate răspândi la orice organ sau țesut, simptomele vor varia. Diverse forme Bolile de la începutul cursului lor sunt caracterizate prin deteriorarea și disfuncția unui organ din corpul uman.
În timp, boala (dacă nu este amiloidoză locală) progresează, afectând alte organe și țesuturi. Manifestările amiloidozei pot fi observate la rinichi, ficat, inimă și glandele suprarenale, splină, tractul gastrointestinal și sistemul nervos, articulații, mușchi și piele. Tipurile de boală sunt descrise în detaliu mai jos.
Leziuni renale
Amiloidoza renală este considerată cea mai mare boala periculoasa, în comparație cu afectarea altor organe. Tabloul clinic al amiloidozei renale depinde de stadiu. În total sunt 4 dintre ele - latente, nefrotice, azotemice, proteinurice.
În stadiul latent, amiloidoza renală nu prezintă practic niciun simptom. Dacă aceasta este o formă secundară, pacientul simte simptomele bolii de bază. Doar ani mai târziu leziunile renale vor deveni simptomatice.
În stadiul proteinuric, amiloidoza renală durează 10 ani sau mai mult. În acest moment, amiloidoza se depune treptat în vasele, spațiul intercelular și glomerulii rinichilor. Din această cauză, nefronii care produc urină sunt comprimați, se atrofiază și mor. Integritatea filtrului renal, care în mod normal nu permite trecerea proteinelor moleculare mari și a celulelor sanguine, este compromisă. Ulterior, proteinele sunt excretate prin urină. În acest stadiu, este dificil să se suspecteze amiloidoza renală, deoarece funcția excretorie nu este afectată. Problema poate fi detectată în rezultatele testelor de laborator.
Amiloidoza renală în stadiul nefrotic se manifestă prin distrugerea ulterioară a filtrului renal. Din această cauză, o cantitate mare de proteine se pierde în urină, iar concentrația acesteia în sânge scade. Proteinele sunt o componentă a procesului de reținere a sângelui în vasele de sânge. Când concentrația de proteine scade, lichidul intră în țesuturi, umflarea apare în orice moment al zilei, indiferent de poziția corpului. În plus, amiloidoza renală progresează, edemul este sever. Lichidul se acumulează în peritoneu, cavitatea pleurală și sacul cardiac. Această etapă durează 4-6 ani.
În stadiul azotemic, din întreg volumul țesut renal doar 25% funcționează. Acest lucru nu este suficient pentru a elimina toxinele dăunătoare, ureea și, prin urmare, concentrația lor crește. Tabloul clinic al insuficienței renale este următorul:
- urinarea este afectată. În loc de 800 ml prescrise pe zi, pacientul excretă mai puțin de 50 ml de urină;
- sănătatea ta se înrăutățește, apar slăbiciune și oboseală;
- digestia este afectată, pofta de mâncare dispare, apar greață și vărsături, gură uscată este însoțită de un miros neplăcut;
- pielea devine palidă, uscată și mâncărime constant;
- sistemul cardiovascular suferă, provocând aritmie, creșterea tensiunii arteriale și posibilă mărire a mușchiului inimii;
- creierul sub influența unei concentrații mari acid uric este deteriorat, apar insomnie și tulburări de memorie, iritabilitate, scăderea abilităților mentale;
- o scădere a hemoglobinei și a globulelor roșii duce la anemie.
Leziuni hepatice
Amiloidoza sistemică se manifestă adesea ca leziuni hepatice. Depozitele de amiloid exercită presiune asupra căilor biliare, vaselor de sânge și celulelor hepatice, ducând la afectarea funcției organelor. Când se disting sindroamele de amiloidoză, ele indică un ficat mărit, resimțit la palpare.
Suprafața ficatului rămâne netedă, nu există durere. În cazul unui curs lung al bolii, rareori se dezvoltă insuficiența hepatică, care este asociată cu abilitățile de regenerare ale organului.
Amiloidoza hepatică se manifestă prin simptome:
- Dimensiunea ficatului crescută.
- Hipertensiunea portală. În mod normal, sângele din organele interne intră în ficat, unde este purificat și apoi returnat în fluxul sanguin. Când vasele hepatice sunt comprimate de amiloid, presiunea în venele organelor interne crește. Ca urmare, apar umflarea picioarelor, diareea cu sânge și sângerarea la nivelul tractului gastrointestinal.
- Icterul apare rar, doar atunci când căile biliare sunt comprimate de depozitele de amiloid. Dacă acesta este motivul, icterul va fi însoțit de mâncărimi ale pielii.
Leziuni cardiace
Amiloidoza cardiacă se dezvoltă în forme primare și alte forme ereditare. Ca urmare a depozitelor de amiloid în miocard și membranele inimii, circulația sângelui este afectată, celule musculare Sunt pe moarte.
Simptomele bolii:
- aritmie;
- cardiomiopatie restrictivă;
- insuficienta cardiaca.
Aritmia apare pe fondul depozitelor de amiloid în mușchiul inimii, care perturbă conducerea impulsurilor nervoase. Ca urmare, camerele inimii se contractă neuniform și apare aritmia. Pacientul se simte amețit și leșină. Din cauza aprovizionării cu sânge a creierului, moartea este posibilă.
Cardiomiopatia restrictivă apare pe fondul depozitelor de amiloid în miocard. Ca urmare, mușchiul inimii se îngroașă și devine mai puțin extensibil, ceea ce duce la funcționarea proastă a camerelor inimii. Tabloul clinic al bolii este oboseală, dificultăți de respirație, o scădere bruscă a tensiunii arteriale în timpul schimbului. pozitie orizontalaîntr-o poziție verticală.
În insuficiența cardiacă, circulația sângelui în organism este perturbată. Acest lucru se manifestă prin umflare și dificultăți de respirație. Insuficiența cardiacă datorată amiloidozei nu răspunde la tratamentul standard pentru bolile cardiovasculare. Boala progresează rapid și duce la moarte în câteva luni.
Leziuni ale glandelor suprarenale și ale splinei
 Glandele suprarenale sunt glande situate pe fiecare rinichi și sunt responsabile de secretarea hormonilor. Amiloidoza perturbă funcția organelor prin oprirea sintezei hormonilor. Dacă amiloidul este depus în splină, organul crește în dimensiune, ceea ce este vizibil la palpare.
Glandele suprarenale sunt glande situate pe fiecare rinichi și sunt responsabile de secretarea hormonilor. Amiloidoza perturbă funcția organelor prin oprirea sintezei hormonilor. Dacă amiloidul este depus în splină, organul crește în dimensiune, ceea ce este vizibil la palpare.
În mod normal, splina elimină celulele deformate din fluxul sanguin care se blochează în structura sa. Depozitele de amiloid fac ca celulele roșii, trombocitele și globulele albe să se blocheze în splină.
Ca urmare, se dezvoltă anemie (slăbiciune generală, piele palidă, dificultăți de respirație), trombocitopenie (sângerări nazale, hemoragii cutanate), leucopenie (sensibilitate la infecții).
Leziuni gastrointestinale
Amiloidoza intestinală poate fi generalizată, atunci când absorbția nutrienților este afectată, și locală, când acumulările de amiloid imită o tumoare. În primul caz, apar simptome precum diaree, pierdere în greutate, slăbiciune, tulburări mentale și anemie. În al doilea caz, boala se caracterizează prin constipație, dureri abdominale și balonare.
Leziuni ale articulațiilor și mușchilor
Amiloidul lovește primul articulații mici pe picioare, pe mâini și, pe măsură ce boala progresează, se instalează în coate și genunchi. Boala se caracterizează prin durere la mișcare, umflarea țesutului și înroșirea pielii, creșterea temperaturii în zona afectată și disfuncția articulației.
Amiloidul se depune neobservat timp indelungat in tesutul conjunctiv, fara a perturba structura muschilor si fara sa apara. De-a lungul timpului, celulele musculare sunt comprimate, alimentarea lor cu sânge este întreruptă și mor. Boala se caracterizează prin slăbiciune musculară, durere, încordare și hipertrofie musculară.
Diagnosticul amiloidozei
Medicii de diferite specializări - reumatologi, cardiologi și urologi, neurologi, dermatologi etc. - pot suspecta un diagnostic precum amiloidoza.De aceea, diagnosticul de amiloidoză ar trebui să se bazeze pe o evaluare cuprinzătoare a istoricului medical, semne clinice, laborator și examen instrumental. Pentru a examina starea organelor, sunt prescrise un ECG, o radiografie a esofagului, endoscopie și sigmoidoscopie. Dacă se suspectează amiloidoza renală, diagnosticul include neapărat o ecografie a cavității abdominale.
Tratamentul amiloidozei
Deși există diferite boală gravă, amiloidoza are un prognostic prost. Faptul este că boala nu poate fi detectată în stadiile incipiente, iar manifestările sale clinice sunt vizibile la mulți ani de la debutul bolii. Cu un diagnostic precum amiloidoza renală, tratamentul este doar de susținere în natură, deoarece măsurile terapeutice nu sunt eficiente.
La prima suspiciune de prezență a bolii este necesară internarea în nefrologie pentru examinare sistemul genito-urinar, deoarece afectarea rinichilor este considerată cea mai mare manifestare periculoasă. Alți specialiști sunt, de asemenea, implicați pentru a examina prezența leziunilor altor organe.
Dacă diagnosticul nu evidențiază tulburări grave în funcționarea vitalului organe importante, tratamentul amiloidozei poate fi efectuat la domiciliu, unde pacientul trebuie să urmeze cu strictețe toate instrucțiunile medicului. Tratamentul poate include medicamente, dietă, dializă și transplant de organe.
„Amiloidoza” este un termen care unește un grup de boli care sunt foarte diverse manifestari cliniceși se caracterizează prin depunerea extracelulară a proteinelor fibrilare patologice insolubile în organe și țesuturi. Această patologie a fost descrisă pentru prima dată în secolul al XVII-lea. Os - splina de sago la un pacient cu abces hepatic. La mijlocul secolului al XIX-lea. Virchow a folosit termenul botanic „amiloid” (din grecescul amylon - amidon) pentru a descrie materialul extracelular găsit în ficat la autopsie, deoarece credea că este similar ca structură cu amidonul. Ulterior, a fost stabilită natura proteică a depozitelor, dar termenul „amiloid” a fost păstrat până în prezent.
În anii 20 În secolul al XX-lea, Benhold a propus colorarea amiloidului cu roșu Congo, apoi a fost descoperit efectul birefringenței în lumina polarizată - o schimbare a culorii roșu cărămidă în verde măr. În 1959, Cohen și Calkins au folosit microscopia electronică pentru a stabili structura fibrilară a amiloidului.
Ideile clinice despre amiloidoză au suferit și ele evoluție: Rokitansky în 1842 a stabilit o legătură între „boala sebacee” și tuberculoză, sifilis și rickettzioză; Wilkes în 1856 a descris „organe grase” la un pacient care nu avea boli concomitente; Atkinson a descoperit amiloidoza la pacienții cu mielom multiplu în 1937. Au fost identificate formele senile (Soyka, 1876) și ereditare (Andrade, 1952), amiloidoza a fost împărțită în genetică, primară și secundară, iar în final, în 1993, a fost adoptată clasificarea OMS, pe baza specificului principalului fibrilar. proteina amiloidului.
În țara noastră, E. M. Tareev, I. E. Tareeva, V. V. Serov au avut o mare contribuție la dezvoltarea ideilor despre amiloidoză. Un rol uriaș în studiul variantelor primare și genetice ale amiloidozei și bolilor periodice îi revine O. M. Vinogradova, ale cărei monografii, publicate în 1973 și 1980, nu și-au pierdut actualitatea astăzi.
În prezent, amiloidoza este împărțită clinic în sistemică și forme locale. Dintre formele sistemice, în funcție de compoziția depozitelor fibrilare, se disting patru tipuri ( ).
Formele locale de amiloidoză includ în prezent boala Alzheimer (A-beta, fibrilele constau din proteina β depusă în creier), amiloidoza insulelor pancreatice, posibil având o legătură patogenetică cu diabetul de tip 2, amiloidoza care apare în tumorile endocrine, tumorile amiloide ale pielea, regiunea nazofaringiană, vezica urinară și alte tipuri rare.
amiloidoza AL
Dezvoltarea amiloidozei AL este posibilă cu mielom, boala Waldenström, limfoame cu celule Bși poate fi idiopatică în amiloidoza primară. Toate aceste opțiuni sunt combinate patogeneza generala, amiloidoza primară este cea mai greu de recunoscut din cauza lipsei semne evidente boala hematologica, prin urmare, merită să ne oprim pe această formă în detaliu.
În amiloidoza primară, discrazie benignă a celulelor plasmatice, înrudite mielom multiplu, clonele anormale ale celulelor plasmatice ale măduvei osoase produc imunoglobuline amiloidogene. Unii aminoacizi din regiunile variabile ale lanțurilor ușoare ale acestor imunoglobuline ocupă o poziție neobișnuită, ceea ce duce la instabilitatea lor și provoacă o tendință de fibrilogeneză. La pacienții cu amiloidoză primară, conținutul de celule plasmatice din măduva osoasă crește la 5-10% (în mod normal, mai puțin de 4%, în mielom - mai mult de 12%) și produc un anumit izotip de lanțuri ușoare de imunoglobuline, care predomină în coloraţia imunohistochimică. Lanțurile ușoare monoclonale libere ale izotipului predominant lambda sau (mai puțin frecvent) kappa sunt detectate în sânge și urină, dar conținutul lor este mai mic decât în mielomul multiplu.
Tabloul clinic al amiloidozei primare este divers și este determinat de implicarea predominantă a anumitor organe în procesul patologic - inima, rinichii, sistemul nervos, tractul gastrointestinal, ficatul etc. Primele simptome sunt slăbiciune și pierdere în greutate, dar în acest sens stadiu, înainte de apariția simptomelor de organ, diagnosticul se pune extrem de rar.
Organele țintă pentru amiloidoza AL sunt cel mai adesea rinichii și inima. Leziunile renale se manifestă prin sindrom nefrotic, care persistă chiar și cu debutul insuficienței renale cronice; hematuria și hipertensiunea arterială nu sunt tipice.
Când amiloidul este depus în miocard, diverse încălcări ritm, insuficiență cardiacă progresivă, care poate fi precedată de modificări asimptomatice ale ECG sub forma unei scăderi a tensiunii undei. Examenul ecocardiografic evidențiază îngroșarea concentrică a pereților ventriculului stâng și drept, o scădere a volumului cavităților inimii, o scădere moderată a fracției de ejecție și disfuncție diastolică a miocardului ventricularului stâng.
Adesea apar simptome de implicare a sistemului nervos - autonom, sub formă hipotensiune arterială ortostatică, și periferice - sub formă de tulburări de sensibilitate. În ultimii ani au început să fie descrise și leziuni ale sistemului nervos central, deși anterior se credea că nu sunt caracteristice amiloidozei primare.
Simptomele dispeptice (senzație de plenitudine, constipație, diaree) și sindromul de malabsorbție pot fi cauzate atât de afectarea sistemului nervos autonom, cât și de amiloidoza tractului gastrointestinal. Hepatomegalia este foarte caracteristică, a cărei natură ar trebui diferențiată între congestie datorată insuficienței cardiace și afectarea ficatului amiloid. Aceasta din urmă este confirmată de o creștere a nivelurilor serice de fosfatază alcalină. Splina este adesea afectată, dar splenomegalia nu este întotdeauna detectată și mare semnificație clinică nu are.
Macroglosia, un semn clasic al amiloidozei primare, se observă la 20% dintre pacienți; infiltrarea țesuturilor moi poate duce la atrofia mușchilor, a pielii, a distrofiei unghiilor, a alopeciei și apariția unor formațiuni asemănătoare tumorii - amiloid.
Mai puțin frecvente sunt leziunile vasculare, ale căror simptome sunt purpura periorbitală - „ochi de raton” și echimoze. Pot apărea sângerări, inclusiv sângerări ale vezicii urinare, cauzate atât de modificări ale peretelui vascular, cât și de o încălcare a sistemului de coagulare, în primul rând o deficiență a factorului X, care se leagă de amiloid. Trombocitoza caracteristică amiloidozei este, de asemenea, explicată de obicei printr-o deficiență a factorilor de coagulare.
Amiloidoza pulmonară este adesea descoperită numai la autopsie. Cu toate acestea, în unele cazuri, dificultățile de respirație, hemoptizia și hidrotoraxul pot fi cauzate nu numai de insuficiența cardiacă congestivă și sindromul nefrotic, ci și de boala pulmonară amiloidă. Este posibilă depunerea de amiloid în alveole și dezvoltarea amiloidului pulmonar. Razele X pot dezvălui modificări reticulare și nodulare ale țesutului pulmonar.
Afectarea glandelor suprarenale poate duce la insuficiență suprarenală, care rămâne adesea nerecunoscută, deoarece hipotensiunea și hiponatremia sunt considerate simptome de insuficiență cardiacă și leziuni ale sistemului nervos autonom. La 10-20% dintre pacienți, hipotiroidismul poate apărea ca o manifestare a leziunii glandei tiroide; se întâlnește adesea mărirea glandelor salivare submandibulare.
Diagnosticul amiloidozei primare, pe lângă caracteristicile clinice indicate, care pot fi similare în amiloidoza secundară, se bazează pe o serie de date de laborator. La 85% dintre pacienți, imunoelectroforeza proteinelor serice și urinare evidențiază imunoglobuline monoclonale. În studiile de rutină, aceleași imunoglobuline monoclonale sunt detectate în urină sub formă de proteină Bence Jones. O biopsie de măduvă osoasă permite diagnostic diferentiat cu mielom multiplu și, de asemenea, relevă o creștere moderată a numărului de celule plasmatice și monoclonalitatea acestora cu colorație imunohistochimică.
Cu toate acestea, chiar și combinația dintre un tablou clinic caracteristic și prezența celulelor plasmatice monoclonale și a proteinelor nu este încă suficientă pentru a confirma diagnosticul de amiloidoză primară. Datele biopsiei joacă un rol decisiv aici. Cea mai puțin invazivă este aspirația țesutului adipos subcutanat al anterioară perete abdominal, oferind 80-90% rezultate pozitive pentru amiloidoza AL (această metodă nu și-a găsit încă aplicație în țara noastră). O biopsie a gingiilor și mucoasei rectale are o anumită valoare diagnostică, dar procentul de rezultate pozitive variază foarte mult, în funcție de stadiul procesului, de aceea este indicat să se efectueze o biopsie a unuia dintre organele afectate - rinichi, ficat. , inima, care da aproape 100% rezultate pozitive pentru amiloidoza de tip AL.
În primul rând, materialul de biopsie este colorat cu roșu Congo. Dacă se detectează congofilia materialului studiat, aceasta trebuie examinată în lumină polarizată; efectul birefringenței este caracteristic doar amiloidului; alte substanțe congofile nu capătă o culoare verde-măr. După aceasta, tiparea amiloid este de dorit. Cea mai precisă este metoda imunohistochimică care utilizează anticorpi monoclonali la proteinele precursoare de amiloid. Cu toate acestea, în prezent este practic indisponibil în țara noastră. Prin urmare, pentru diagnostic, se folosește colorarea cu soluții de guanidină alcalină sau permanganat de potasiu, ceea ce permite, deși indirect, determinarea tipului de depozite fibrilare.
Prognosticul pentru amiloidoza primară este mai rău decât pentru alte forme de boală, durata medie viața nu depășește doi ani; în prezența leziunilor cardiace sau a leziunilor multisistem fără tratament, pacienții mor în câteva luni. Cel mai motive comune decesele sunt insuficiența cardiacă și renală, sepsisul, complicațiile vasculare și cașexia. Similitudinea patogenetică cu mielomul multiplu permite să ne așteptăm la inhibarea progresiei bolii cu chimioterapie administrată pentru suprimarea celulelor plasmatice monoclonale. Există mai multe regimuri de tratament ().
Utilizarea chimioterapiei, dacă tratamentul are succes, poate crește speranța de viață a pacienților cu 10 până la 18 luni. Dar eficacitatea terapiei este scăzută, în special datorită faptului că, în multe cazuri, progresia bolii duce la moartea pacienților înainte de finalizarea cursului de tratament, precum și datorită dezvoltării citopeniei, complicatii infectioase, aritmii fatale în timpul tratamentului cu doze ultra-mari de dexazonă. Utilizarea de doze mari de melfolan cu transplant de celule stem autologe permite obținerea remisiunii în mai mult de 50% din cazuri, cu toate acestea, utilizarea acestei metode este limitată de severitatea afecțiunii, vârsta pacienților, tulburări funcționale din inimă și rinichi. În multe cazuri, este posibilă doar terapia de întreținere simptomatică.
amiloidoza AA
Dezvoltarea amiloidozei AA are loc în timpul proceselor inflamatorii cronice, precursorii amiloidului AA sunt proteinele serice de fază acută, α-globulinele produse de celule. tipuri diferite, în principal neutrofile și fibroblaste. Amiloidoza secundară se dezvoltă cu poliartrită reumatoidă, spondilită anchilozantă, artrită psoriazică, diverse tumori, limfogranulomatoză, colită ulceroasă și boala Crohn, cu boală periodică (febră mediteraneană familială), precum și cu tuberculoză, osteomielite, bronșiectazie.
Trăsăturile clinice caracteristice ale amiloidozei AA sunt afectarea rinichilor la majoritatea pacienților, precum și afectarea relativ rară a ficatului și/sau splinei (aproximativ 10%) și a inimii (detectată numai prin ecocardiografie). Macroglosia nu este tipică pentru amiloidoza secundară. Diagnosticul se bazează pe o combinație de amiloidoză renală și boală inflamatorie cronică, confirmată prin colorarea imunohistochimică a materialului de biopsie; în țara noastră se folosesc metodele de colorare indirectă deja menționate mai sus.
Prognosticul depinde în mare măsură de natura bolii de bază; în cursul natural, o treime dintre pacienți dezvoltă insuficiență renală la 5 ani de la momentul în care este detectată proteinuria. Pentru bolile periodice, rata de supraviețuire la cinci ani este de 25%.
Tratamentul se bazează pe suprimarea focalizării - sursa de producere a proteinelor precursoare ale serului. Îndepărtarea tumorilor, sechestrectomia, rezecția intestinală, tratamentul tuberculozei, reducerea activității artritei reumatoide (cu utilizarea de citostatice) duc la încetarea progresiei amiloidozei și, uneori, la dezvoltarea inversă a manifestărilor clinice, în special nefrotice. sindrom.
Utilizarea colchicinei pentru boli periodice este metoda de alegere, eficacitatea sa a fost dovedită, tratamentul previne dezvoltarea amiloidozei și inhibă progresia acesteia. În alte forme de amiloidoză secundară, eficacitatea colchicinei nu a fost confirmată.
Formele senile și ereditare de amiloidoză sistemică, precum și formele locale, sunt rare; amiloidoza de dializă este bine cunoscută specialiștilor și practic nu este niciodată întâlnită în practica generală.
Terapia simptomatică nu depinde de tipul de amiloidoză, ci de organele țintă afectate ( ).
Amiloidoza, în special primară, este considerată o patologie neobișnuită, dar în realitate nu este atât de rară, încât este dificil de diagnosticat. Diagnosticul adecvat necesită nu numai cunoașterea tabloului clinic și patogeneza bolii, ci și disponibilitatea anumitor capacități de diagnosticare. Pentru a ilustra acest punct, prezentăm propriile noastre date ( ). În secția de nefrologie a Spitalului Clinic Orășenesc S.P. Botkin din Moscova în anii 1993-2003. Au fost observați 88 de pacienți care au fost diagnosticați cu amiloidoză.
Diagnosticul a fost confirmat morfologic la toți pacienții cu amiloidoză AL, amiloidoză senilă și neprecizată, iar la 30 de pacienți cu amiloidoză secundară - în total 53 de cazuri. La 12 pacienți s-a efectuat o biopsie renală, la doi pacienți s-a efectuat o biopsie hepatică, la 8 pacienți s-a efectuat o biopsie intestinală, în 12 cazuri s-a efectuat o biopsie a gingiilor, iar în alte 19 cazuri s-a confirmat diagnosticul prin examen morfologic de secțiune. material.
În cele mai multe cazuri, diagnosticul de amiloidoză a fost pus pentru prima dată ca urmare a examinării în secția de nefrologie. Am făcut o comparație între pacienții cu amiloidoză AL de referință și diagnostice clinice ( ).
În doar două cazuri din 20 (10%) diagnosticul de trimitere a fost „amiloidoză primară”, iar unul dintre acești pacienți a fost diagnosticat la clinica de terapie MMA și boli profesionale, iar celălalt la o clinică străină.
Toți pacienții care au fost diagnosticați cu mielom cu dezvoltarea amiloidozei AL au fost transferați la secțiile de hematologie. Din cei 11 pacienți cu amiloidoză primară, șapte pacienți au primit chimioterapie cu o combinație de melfolan și prednisolon oral în cure intermitente, patru dintre ei - în combinație cu tratamentul de dializă și un alt pacient - doar dializă și tratament simptomatic. Dintre acești pacienți, cinci persoane au murit într-o perioadă de două săptămâni până la doi ani de la începutul tratamentului (toți cu insuficiență renală și leziuni de organe multiple), un pacient este sub dializă, un pacient a fost îndrumat pentru transplant autologe de celule stem și unul. pacientul primește tratament până în prezent. La un pacient, chimioterapia a fost amânată din cauza prezenței unui ulcer gastric de lungă durată, iar alți doi pacienți au refuzat tratamentul.
Dintre pacienții cu amiloidoză secundară din studiul nostru, au predominat pacienții cu poliartrită reumatoidă; osteomielita cronică și artrita psoriazică au fost pe locul doi în rândul cauzelor; alte boli au fost mai puțin frecvente ( ).
Tratamentul poliartritei reumatoide și al artritei psoriazice a fost efectuat cu utilizarea de citostatice (metatrexat, azatioprină), deși în multe cazuri opțiunile de tratament au fost limitate din cauza prezenței insuficienței renale cronice și a patologiei concomitente. Pacienții cu osteomielita cronică au fost trimiși în secții de chirurgie purulentă. Pacienții cu spondilită anchilozantă și boala Crohn au primit tratament specific, pacienții cu BPOC și tuberculoză au fost trimiși și în spitale de specialitate. Unul dintre pacienții cu tumoră la stomac a fost operat cu succes, iar pe parcursul a patru ani de observație, sindromul nefrotic a regresat treptat; în alte cazuri de tumori, prevalența procesului a permis doar terapie simptomatică; pacientul cu limfogranulomatoză a fost internat. la starea terminală. Rata mortalității în rândul pacienților cu amiloidoză secundară a fost de 38% (datorită pacienților cu leziuni avansate la momentul diagnosticului). Toți pacienții cu boli periodice au primit terapie cu colchicină.
Caracteristicile diagnosticului și aplicării metodelor moderne de tratament al amiloidozei primare pot fi ilustrate prin următorul exemplu: pacientul K., în vârstă de 46 de ani, a fost internat pentru prima dată la sfârșitul lunii octombrie 2002 cu plângeri de umflare a picioarelor, palpitații și amenoree. În anamneză - raceli, apendicectomie, două nașteri normale de urgență, fără indicații de boală renală, fără boli cronice. În aprilie 2002 s-a transferat pneumonie acutăîn lobul superior al plămânului drept, a fost tratat în ambulatoriu, a primit injecții cu abactal și lincomicină. Din cauza localizării pneumoniei, a fost examinată la o clinică de tuberculoză, iar diagnosticul de tuberculoză a fost exclus. La începutul lunii iunie, a apărut pentru prima dată umflarea picioarelor, pentru care nu a fost examinată. Umflarea prin un timp scurt au fost lichidate pe cont propriu, apoi au fost reluate. Pacientul a fost internat într-un spital terapeutic; examenul a evidențiat proteinurie de până la 1,65%, hipoproteinemie (proteine serice totale 52 g/l), tensiune arterială normală (120/80 mm Hg), sediment urinar nemodificat, valorile plasmatice ale creatininei sunt de asemenea în limite normale. limite. S-a pus diagnosticul de glomerulonefrită acută, s-a efectuat tratament cu ampicilină, clopoțel, heparină, triampur și s-a efectuat o amigdalectomie. Proteinuria a persistat, edemul a crescut treptat și, prin urmare, pentru examinare și tratament ulterioară, pacientul cu diagnostic de glomerulonefrită cronică a fost trimis la spitalul numit după. S. P. Botkina.
La examinare pielea este curata, de culoare normala, anasarca, edem masiv, dens, se detecteaza ascita, ganglionii limfatici periferici nu sunt mariti. Tensiune arterială 110/70 mm Hg. Art., zgomotele cardiace sunt sonore, clare, ritmice, frecvența cardiacă 90 bătăi/min, ficatul și splina nu sunt mărite, diureza este de până la 1000 ml/zi, scaunul este regulat, fără impurități patologice. Examenul a evidențiat sindrom nefrotic - proteinurie 3 g/l, sediment urinar slab, hipodisproteinemie, hiperlipidemie (proteine serice totale 39 g/l, albumină 12 g/l, globuline 7-30-15-19%, respectiv α 1 -α 2 -β-γ colesterol 17,8 mmol/l, β-lipoproteine 250 UI), la analiza urinei pentru proteina Bence-Jones - reacția este negativă, excreția zilnică de 17-KS nu este redusă. Testul clinic de sânge și alți parametri biochimici sunt în limite normale, coagulograma arată hiperfibrinogenemie severă, nivel crescut de RKFM. Studiul imunoglobulinelor din sânge: Ig-A - 0,35, Ig-M - 35,7 (două norme), Ig-G - 1,96 g/l. Radiografie a organelor toracice, craniu și oase pelvine, ecografie a cavității abdominale, rinichi, glanda tiroidă, ECHO-CG fără patologie, ecografie pelviană - semne de adenomioză a corpului uterin, endoscopie - esofagită de reflux, gastrită cronică. Când a fost examinat de un neurolog, nu a fost găsită nicio patologie; oncologul a diagnosticat mastopatie fibrochistică.
Pentru a clarifica geneza sindromului nefrotic, s-a efectuat o biopsie prin puncție cu ac fin a rinichiului drept sub anestezie locală și ghidaj ecografic, fără complicații. La examinarea unei probe de biopsie, se observă depunerea de amiloid în mezangiu glomerular și în vasele extraglomerulare. Amiloid încarcă până la 25% din ansele vasculare glomerulare. Un studiu imunohistochimic nu a găsit nicio luminiscență specifică. Când preparatele sunt tratate cu o soluție alcalină de guanidină timp de 2 ore, congofilia și proprietățile lor în lumină polarizată sunt păstrate, ceea ce este caracteristic amiloidozei AL.
Pentru a clarifica natura amiloidozei AL, în laboratorul Immunotest a fost efectuat un studiu imunochimic al sângelui și urinei. Au fost detectate paraproteinemia M-lambda cu o scădere a nivelului de imunoglobuline policlonale și paraproteinurie de tip lambda Bence-Jones pe fondul proteinuriei masive neselective. Pacientul a fost consultat de un hematolog, s-a sugerat prezența bolii Waldenström și s-a efectuat o biopsie de măduvă osoasă. Concluzie: în cavitățile existente ale măduvei osoase sunt vizibile celulele tuturor celor trei germeni ai hematopoiezei normale, precum și celulele limfoide care nu formează grupuri. Diagnosticul bolii Waldenström a fost respins din cauza absenței infiltrației limfoide a măduvei osoase, a măririi ganglionilor limfatici și a splinei și a absenței substratului tumoral.
S-a stabilit un diagnostic de amiloidoză primară cu afectare renală, sindrom nefrotic, funcție renală conservată; nu au fost detectate semne de afectare a altor organe. În ianuarie 2003 a fost începută chimioterapia cu melfolan 16 mg/zi și prednisolon 100 mg/zi, cure de patru zile la fiecare șase săptămâni. Se efectuează și tratament simptomatic: furosemid, veroshpiron, preparate cu potasiu, famotidină, transfuzii cu albumină. Până în prezent, au fost efectuate cinci cure de chimioterapie cu toleranță bună, edemul a scăzut, proteinuria a scăzut la 1,8 g/l, iar severitatea hipodisproteinemiei a scăzut ușor (proteine totale 46 g/l, albumină 18 g/l, α 2 -globuline 20%). Funcția rinichilor rămâne intactă, creatinina plasmatică este de 1,3 mg/dL și nu au fost detectate semne de afectare a altor organe și sisteme în timpul examinărilor de control dinamic.
Acest caz ilustrează clar faptul că examenul morfologic, imunologic și imunochimic este necesar pentru a diagnostica amiloidoza. Astfel, la pacientul nostru, cel mai evident diagnostic clinic a fost „glomerulonefrita cronică”, iar în absența posibilității de a efectua o biopsie renală, cel mai probabil acest diagnostic ar fi fost pus. Fără indicații clinice de natură sistemică a bolii, cronice proces inflamator, pacientul nu a avut nicio boală a sistemului sanguin, cu excepția unei creșteri a nivelului de Ig-M. Și numai datele obținute din studiul biopsiei renale au implicat o biopsie a trepanei măduvei osoase și un studiu imunochimic, care împreună au făcut posibilă stabilirea unui diagnostic de amiloidoză primară înainte de apariția leziunilor sistemice. Terapia patogenetică a fost început, deși pe fondul sindromului nefrotic deja dezvoltat, dar înainte de debutul insuficienței renale și când doar 25% din glomeruli erau încărcați cu amiloid, care are un prognostic relativ favorabil.
În concluzie, observăm că amiloidoza este boala grava Cu nivel inalt mortalitatea, care este extrem de dificil de diagnosticat, dar examinarea la timp și de înaltă calitate a pacienților face posibilă stabilirea unui diagnostic mai devreme și numirea la timp terapia adecvată, la rândul său, face posibilă îmbunătățirea prognosticului la acest grup de pacienți.
Literatură
- Varshavsky V. A., Proskurneva E. P. Semnificația și metodele de diagnostic morfologic al amiloidozei în Medicină modernă// Nefrologie practică. - 1998. - 2:16-23.
- Vinogradova O. M. Variante primare și genetice ale amiloidozei. - M.: Medicină, 1980.
- Zakharova E. V., Khrykina A. V., Proskurneva E. P., Varshavsky V. A. Un caz de amiloidoză primară: dificultăți în diagnostic și tratament // Nefrologie și dializă. - 2002. - 1:54-61.
- Rameev V.V. Caracteristici ale afectarii rinichilor in amiloidoza AA si AL: diss... cand. Miere. Sci. - M., 2003.
- Kozlovskaya L.V., Varshavsky V.A., Chegaeva T.V. și colab. Amiloidoza: aspect modern la problema // Nefrologie practică. - 1998. - 2:24-26.
- Rodney H., Raymond L.C. și Skinner M. // Amiloidozele sistemice; New England Journal of Medicine, 1997. - 337:898-909.
- Dhodapkar M.V., Jagannath S., Vesole d. et al // Tratamentul AL-amiloidozei cu dexametazonă plus interferon alfa / Limfom Leuc. 1997. - 27(3-4):351-365
- Gertz M.A., Lacy M.Q., Lust J.A. et all // Studiu de fază II cu dexametazonă în doze mari pentru amiloidoza cu lanț ușor cu imunoglobuline tratată anterior. Am J Hematol 1999,61(2):115-119.
- Gertz M.A., Lacy M.,Q., Lust J.A. et al // Studiu de fază II cu dexametazonă în doză mare la pacienții netratați cu amiloidoză sistenică primară. Med Oncol 1999.- 16(2):104-109
- Sezer O., Schmid P., Shweigert M. și colab. // Inversarea rapidă a sindromului nefrotic din cauza amiloidozei AL sistemice primare după VAD și chimioterapie ulterioară cu doze mari cu suport de vânzare tulpină autologă. Transplant de măduvă osoasă. 1999. - 23(9): 967-969.
- Sezer O., Neimoller K., Jakob C. et al // Abordări noi pentru tratamentul amiloidozei primare. Expert Opin Investig Grugs. 2000. - 9(10):2343-2350
- Sezer O., Eucker J., Jakob C., Possinger K. // Diagnosticul și tratamentul amiloidozei AL. Clin Nephrol. 2000. - 53(6):417-423.
- Skinner M. Terapia curentă „Amyloidosis” în alergie, imunologie și reumatologie. Cartea Anului Mosby. 1996. - 235-240.
- Palladini G., Anesi E., Perfetti V. et al. Un regim modificat cu doze mari de dexametazonă pentru amiloidoza sistemică primară (AL). Jurnalul Britanic de Hematologie. 2001. - 113:1044-1046.
E. V. Zakharova
Orașul Moscova Spitalul clinic lor. S. P. Botkina
Tabelul 2. Regimuri de tratament pentru amiloidoza primară
- Administrare orală ciclică de melfolan (0,15-0,25 mg/kg greutate corporală pe zi) și prednisolon (1,5-2,0 mg/kg pe zi) timp de patru până la șapte zile la fiecare patru până la șase săptămâni timp de un an, până la atingerea dozei de curs de 600 mg
- Administrare orală melfolan în doză de 4 mg/zi timp de trei săptămâni, apoi, după o pauză de două săptămâni - 2-4 mg/zi patru zile pe săptămână continuu, până la atingerea dozei de curs de 600 mg, în asociere cu prednison
- Administrarea intravenoasă de doze mari de melfolan (100-200 mg/m² suprafață corporală timp de două zile) urmată de transplant autologe de celule stem
- Dexametazonă intravenoasă 40 mg timp de patru zile la fiecare trei săptămâni - opt cicluri
- Administrarea intravenoasă de dexametazonă în doză de 40 mg în prima zi a patra, a 9-12 și a 17-20 a unui ciclu de 35 de zile, trei până la șase cicluri, urmată de utilizarea a-interferonului în doză de 3- 6 milioane de unități de trei ori pe zi pe săptămână
- Regimul vincristină-doxoribucin-dexametazonă (VAD).
