Defekte dhe anomali kongjenitale. Defektet e lindjes: shkaqet, parandalimi dhe trajtimi
Disa nga defektet e lindjes më poshtë janë mjaft të njohura, ndërsa të tjerat janë jashtëzakonisht të rralla. Por sido që të jetë, ato janë të gjitha mjaft rrëqethëse, misterioze dhe tragjike. Kështu që…
Kjo është një gjendje e rrallë (rreth një në 200,000 lindje) në të cilën binjakët lindin të bashkuar në një ose më shumë pjesë të trupit. Në 70-75% të të gjitha rasteve, binjakët siamez janë femra. Rreth gjysma kanë lindur të vdekur. Ndonjëherë ato mund të ndahen, gjë që lejon Binjakët siamezë jetojnë jetë e plotë por më shpesh kjo nuk është e mundur.
Hipertrikoza (sindroma Ambrams)
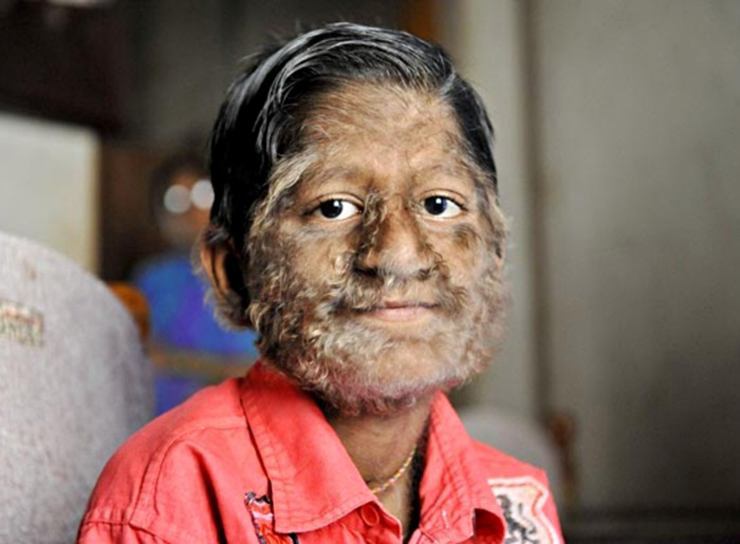
Hipertrikoza është një sëmundje e karakterizuar nga rritje e tepërt qime, të pazakonta për këtë zonë të lëkurës. Për fat të mirë, kjo është shumë, dhe aktualisht ka vetëm 40 njerëz në botë që vuajnë nga hipertrikoza. Sëmundja është jashtëzakonisht dobësuese për fëmijët, pasi ata shpesh refuzohen nga bashkëmoshatarët e tyre.
Sirenomelia (sindroma e sirenës)
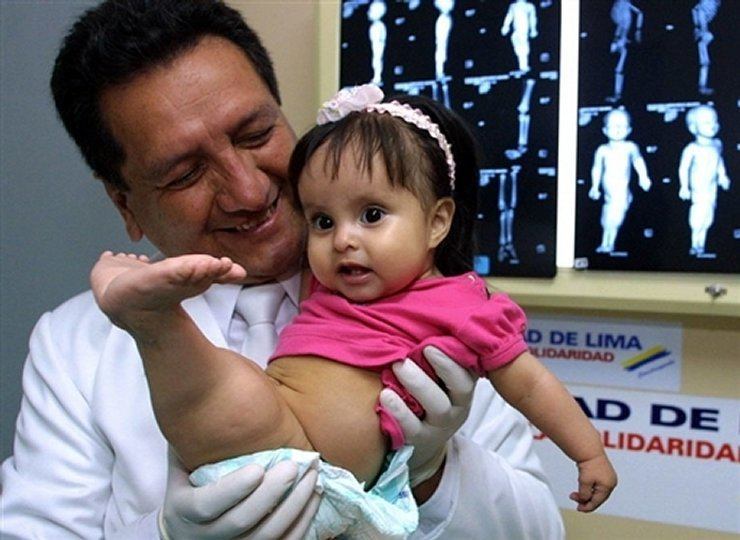
Sirenomelia është një anomali zhvillimore që manifestohet në formën e shkrirjes ekstremitetet e poshtme. Ndodh në një rast për 100 mijë të porsalindur. Si rregull, çon në vdekje, 1-2 ditë pas lindjes, kjo është për shkak të çuditshmërive në zhvillimin dhe funksionimin e veshkave dhe Vezika urinare. Megjithatë, ka raste kur fëmijët me këtë anomali (edhe pa ndërhyrje kirurgjikale) jetoi për disa vjet. Pra, vajza amerikane Shiloh Pepin, e cila vuante nga sirenomelia, mundi të jetonte për më shumë se 10 vjet.
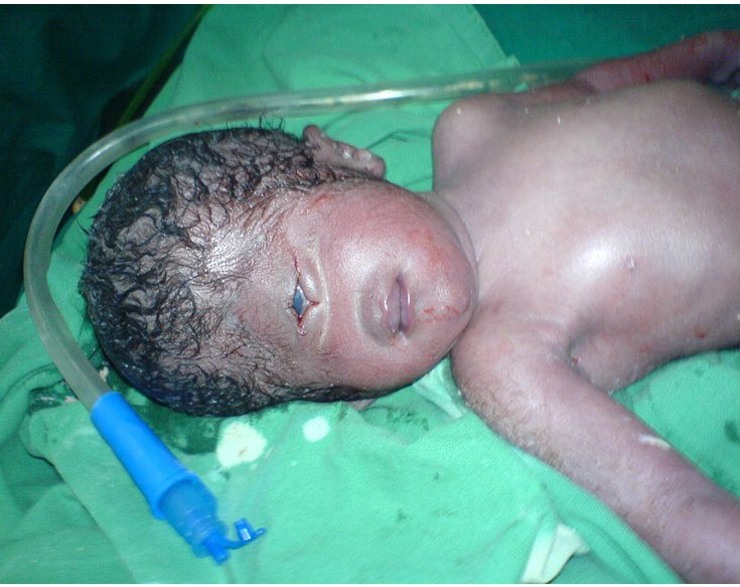
Cyclopia, siç mund ta merrni me mend, është emëruar pas krijesës së famshme mitike Cyclops. Fëmijët e lindur me ciklopi kanë vetëm një sy, të vendosur në mes të kokës. Në 100% të të gjitha rasteve, të porsalindurit vdesin në ditët e para të jetës.
Lloji i shkrirjes së binjakëve, në të cilin në kokë fëmijë normal koka e binjakut, e cila nuk ka trup, rritet. Historia njeh vetëm dhjetë shembuj të regjistruar të kësaj anomalie dhe vetëm në tre prej tyre fëmija mbeti gjallë pas lindjes. Në një rast, koka e dytë ishte në gjendje të buzëqeshte, të mbyllte sytë, të qante dhe të thithte gjoksin e nënës.
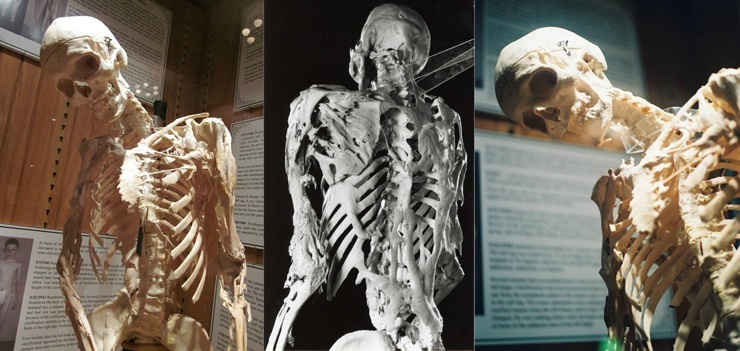
Sëmundja më e rrallë(1 rast në 2 milion), që rezulton nga një mutacion i gjeneve dhe i manifestuar nga defekte kongjenitale zhvillimore - kryesisht të lakuara gishtat e mëdhenj ndalesë dhe shkelje në rajoni i qafës së mitrës shtylla kurrizore. Baza e fibrodisplazisë është formimi proceset inflamatore në tendinat, ligamentet, fascinë, aponeurozat dhe muskujt, të cilat në rezultati përfundimtarçon në kalcifikimin dhe kockëzimin e tyre. Sëmundja quhet ndryshe edhe “Sëmundja e skeletit të dytë”, pasi në fakt aty ku në organizëm kërkohen procese të rregullta antiinflamatore, fillon rritja e kockave.
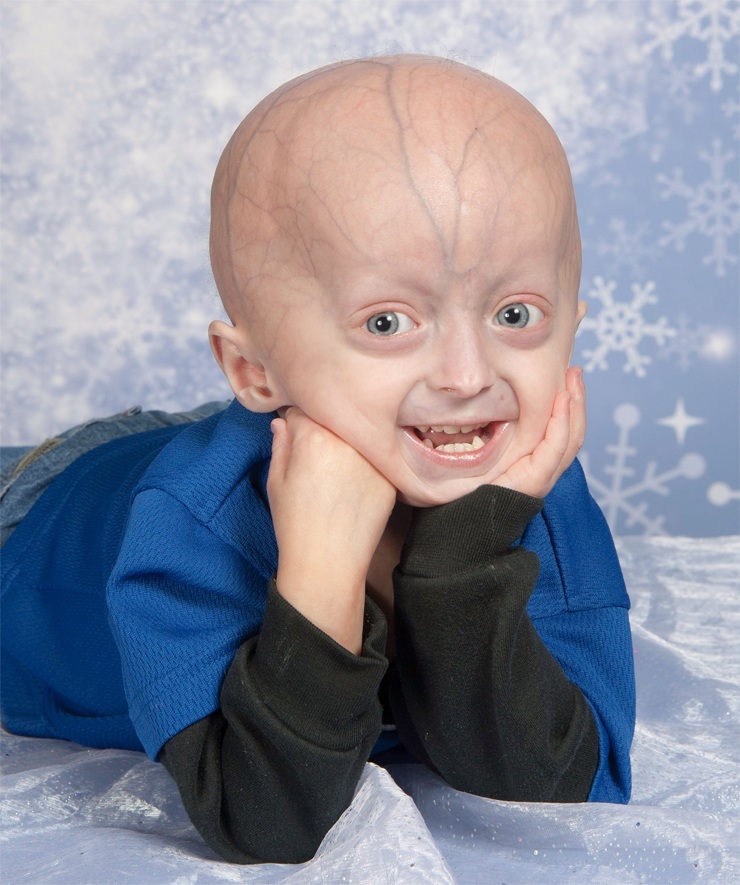
Progeria - më e rralla defekt gjenetik në të cilat ndodhin ndryshimet e lëkurës dhe organet e brendshme, për shkak të plakja e hershme organizëm. Në botë nuk janë regjistruar më shumë se 80 raste të progerisë.
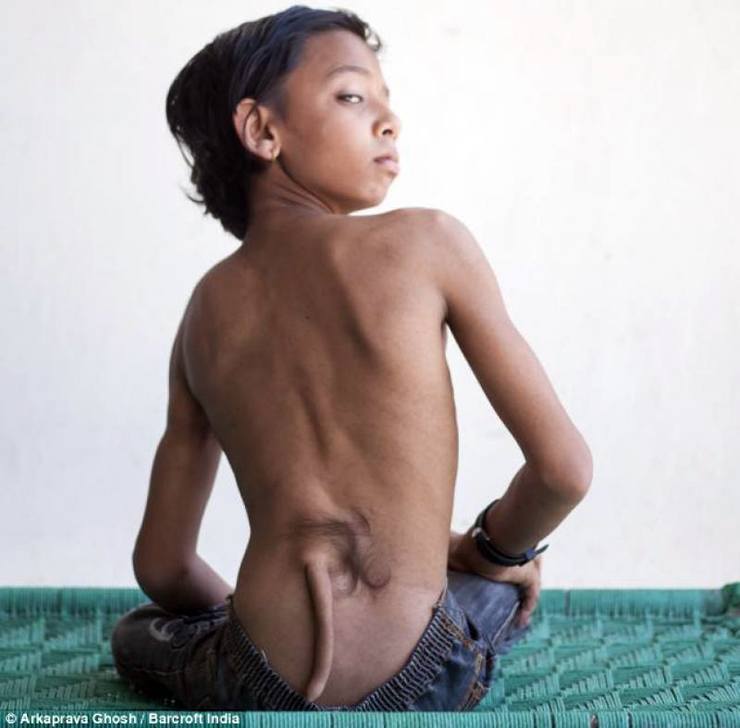
defekt i lindjes, në të cilën foshnja lind me një bisht gjysmë funksional, të kompletuar me muskuj, nerva, lëkurë dhe enët e gjakut. Mendohet se shkaktohet nga një mutacion gjenetik.

Anencefalia - e plotë ose mungesa e pjesshme hemisferat truri, kockat e kafkës dhe indet e buta. Ndodh afërsisht një herë në 10 mijë të porsalindur (në SHBA), më shpesh tek fetuset femra. Defekti në 100% të rasteve është vdekjeprurës. 50% e fetuseve me anencefali vdesin në mitër, 50% e mbetur lindin të gjallë, por vetëm 66% mund të zgjasin disa orë (megjithatë, ka raste që disa kanë jetuar për rreth një javë). Stephanie Keane, e njohur më mirë me pseudonimin Baby Kay, konsiderohet si "mëlçia e gjatë" në mesin e anencefalianëve, të cilët jetuan me këtë diagnozë të tmerrshme për 2 vjet e 174 ditë.
Shpërndaje në rrjetet sociale rrjetet
Përafërsisht 2-3% e të porsalindurve kanë keqformime të rënda kongjenitale. Embriologjikisht, defekte të tilla klasifikohen në tre klasa kryesore (Tabela 36-6):
defekte te lindjes si rezultat i morfogjenezës jo të plotë;
Defektet e lindjes si rezultat i morfogjenezës së përsëritur;
Defektet e lindjes si rezultat i morfogjenezës aberrante. Morfogjeneza jo e plotë është patologjia më e zakonshme, aberrante - më e rralla.
Tabela 36-6.
(Cohen M.M., 1997)
Pothuajse të gjitha keqformimet kongjenitale ndodhin në periudha embrionale(java 3-10 e shtatzënisë), gjatë periudhës kur ndodh diferencimi i organeve (Tabela 36-7).
Defektet e lindjes mund të jenë të thjeshta ose komplekse. Si afati i mëvonshëm shfaqja e një defekti të lindur, më shumë gjasa se nuk do të ketë defekte të lidhura patogjenetikisht në strukturat embrionale fqinje (keqformime të thjeshta kongjenitale). Nëse një defekt shfaqet në fazat e hershme të embriogjenezës, probabiliteti i përfshirjes së strukturave ngjitur është mjaft i lartë dhe një kaskadë e shumëfishtë defekte te lindjes zhvillimi ose sekuenca (sekuenca) e keqformimeve kongjenitale. Një shembull është sekuenca e Pierre Robin, kur defekti parësor në formën e hipoplazisë intrauterine mandibulë shkakton një shkelje të procesit të uljes së gjuhës, e cila, nga ana tjetër, çon në mosmbylljen e qiellzës.
Tabela 36-7. Koha e formimit të keqformimeve kongjenitale (Cohen M.M., 1997)
keqformime kongjenitale në të manifestimi klinik mund të jetë minimale (bifurkacioni i uvulës) dhe maksimalisht i theksuar (mosokluzioni i qiellzës). Në rastin e manifestimit minimal, do të përkufizohet si anomali e vogël zhvillimin. Një keqformim ose sekuencë komplekse mund të paraqitet gjithashtu në minimum variant klinik.
Kështu, për shembull, sekuenca e holoprosencefalisë në versionin e saj më të rëndë karakterizohet nga një keqformim kongjenital i hemisferave cerebrale dhe anomalive të fytyrës - mungesa e strukturave të hundës, hipotelorizmi, ageneza premaksilare me buzë të çara dhe procesi alveolar nofullën e sipërme; në një variant minimal klinik, karakterizohet nga një kombinim i hipotelorizmit me një incizues të vetëm maksilar. Njohja e kësaj është jashtëzakonisht e rëndësishme për prognozën gjenetike familjare të një fëmije me holoprosencefali - ekzaminimi i prindërve për manifestime klinike minimale.
DEFORMIMET
Ky lloj defekti i lindjes gjendet në afërsisht 1-2% të të porsalindurve. Defektet më të shpeshta janë këmba e shtruar, dislokimi kongjenital i ijeve dhe skolioza pozicionale (skolioza posturale). Deformimet më së shpeshti ndodhin në periudhën e vonë të fetusit si pasojë e tre shkaqeve kryesore dhe faktorëve predispozues (Tabela 36-8): shkaqet mekanike; keqformime kongjenitale; arsye funksionale.
Shkaqet mekanike të deformimeve janë më të zakonshmet dhe ndodhin në sfondin e hipokinezisë fetale. Në një studim të kryer me 4500 të porsalindur, u tregua se në mesin e të gjithë të porsalindurve me deformime, 1/3 e fëmijëve kishin dy ose më shumë deformime. Kjo sekuencë deformime kongjenitale ilustrohet mirë nga një shembull ku ngurtësia e mitrës është shkaku i tre deformimeve - plagiocefalisë, asimetrisë së nofullës së poshtme dhe shputës së shtruar në një të porsalindur.
Tabela 36-8. Faktorët predispozues në zhvillimin e deformimeve (Cohen M.M., 1997)
| Mekanike | |
| shkaqet | Ngurtësia e mitrës dhe muskujve të barkut (e lidhur me |
| lindja e parë) | |
| Shtat i shkurtër dhe përmasa e zvogëluar e trupit të një gruaje shtatzënë | |
| femrat | |
| hipoplazia unazë pelvike | |
| Hipoplazia e mitrës | |
| uterus dycornuate | |
| Leiomyoma e mitrës | |
| Vend i pazakontë i implantimit | |
| Rrjedhje kronike e lëngut amniotik | |
| oligohidramnios ( etiologji të ndryshme) | |
| Pozicioni i pazakontë i fetusit | |
| Futja e hershme e legenit të kokës së fetusit | |
| Shtatzënia e shumëfishtë | |
| Keqformime kongjenitale të fetusit | |
| fruta të mëdha(makrosomia kongjenitale) | |
| Makrocefalia ose hidrocefalusi fetal | |
| defekte te lindjes | Spina bifida; |
| Zhvillimi i fetusit si | |
| shkak i deformimeve | Keqformime të tjera kongjenitale sistemi nervor fetusit |
| Agjeneza e veshkave fetale (dypalëshe) | |
| Hipoplazia e rëndë renale | |
| Sëmundja e rëndë e veshkave policistike | |
| Atrezi uretral | |
| Funksionale | Çrregullime neurologjike(hipotensioni kongjenital) |
| shkaqet | |
| Çrregullime muskulare | |
| Defektet IND lidhës | |
Pothuajse të gjitha defektet e rënda të lindjes sistemi urinar shkaktojnë oligohydramnios, i cili, nga ana tjetër, është shkaku i sindromës Potter (fytyrë e pazakontë e fetusit, kontraktime të shumta të gjymtyrëve).
Shkaqet funksionale të deformimeve përfshijnë forma të ndryshme hipotensioni kongjenital i të porsalindurve dhe llojet neuromuskulare të artrogripozës. Hipotensioni kongjenital mund të shoqërohet me mikrognatia, mikroglosia, sutura anësore të zgjatura qiellza e fortë, palosjet jonormale të përkuljes së dorës dhe këmbës, sheshtë-valgus këmbë dhe deformime të tjera. Artrogripoza karakterizohet nga ngurtësi kongjenitale e gjymtyrëve dhe fiksim i kyçeve në një pozicion karakteristik.
ÇRREGULLIMI
Frekuenca e saktë e çrregullimeve nuk dihet, zbulohet në 1-2% të të porsalindurve. Studiuesi i parë që përshkroi kjo specie patologjisë në monografinë e vitit 1968 "Fetal Malformations Caused by Amnion Rupture Gjatë Gestation", ishte R. Torpin (Cohen M.M., 1997). Çrregullimet lindin si rezultat i ekspozimit arsye të ndryshme: faktorët vaskular, anoksia, infeksionet, rrezatimi, teratogjenët, brezat amniotikë, faktorët mekanikë. Lloji dhe ashpërsia e ndërprerjeve varen nga mosha gestacionale, vendndodhja e ndikimit dhe shkalla e dëmtimit të indeve. Më shpesh, ndërprerjet ndodhin gjatë periudhës së fetusit, megjithatë, efektet teratogjene janë karakteristike për morfogjenezën embrionale. Disa nga këto ekspozime, që ndodhin gjatë periudhës embrionale, “fenokopojnë” keqformimet kongjenitale. Për shembull, shiritat amniotikë afati i hershëm shtatzënitë mund të shkaktojnë anencefalinë, çarje të buzës dhe qiellzës, defekte të reduktimit të ekstremiteteve. me e veshtira diagnoza diferenciale ndërmjet keqformimit kongjenital dhe ndërprerjes si pasojë e patologjisë vaskulare (Tabela 36-9).
Tabela 36-9. Mekanizmat e përçarjes vaskulare në embrion dhe fetus (Cohen M.M., 1997)
| Patogjeneza | Anomali strukturore |
| Shkatërrimi i embrionit rrjeti kapilar | Sekuencimi i hershëm amniotik, kompleksi i murit gjymtyrë-bust, anomalitë e reduktimit të gjymtyrëve, hipoplazia maksilare dhe e gjymtyrëve |
| Qëndrueshmëria e enëve embrionale | Defektet e gjymtyrëve: aplazia radiale, aplazia tibiale, aplazia fibulare, shputa e shtruar |
| Amputimi i parakohshëm i enëve embrionale | Sekuenca e patologjisë arteria subklaviane(Sekuencat Poloni, Möbius, Klippel-Feil), gastroschisis, veshka patkua |
| Shkelja e maturimit vaskular | Hemangiomat kapilare, fistulat arteriovenoze, aneurizmat (aneurizmat Berry) |
| Mbyllja (ngjeshja e jashtme) e enëve të gjakut | Anomalitë e shoqëruara me leiomioma, me shtatzënia tubale dhe uterus dycornuate |
| Mbyllja (tromboza embolike) e enëve të gjakut | Porencefalia, hydranencefalia, mikrocefalia, atrezia e fshikëzës së tëmthit, sindaktilia distale, mikrosomia hemifaciale (rrallë), anorkizmi dypalësh, aplazia e lëkurës |
| Shkelja e hemodinamikës | Anomalitë e shkaktuara nga përdorimi i kokainës gjatë shtatzënisë |
Defektet e izoluara kongjenitale të zhvillimit nuk shkaktojnë vështirësi në diagnostikim. Një situatë krejtësisht tjetër vërehet në fushën e defekteve të shumëfishta të lindjes, ku përvoja dhe njohuritë empirike për diagnostikimin dhe trajtimin e fëmijëve me defekte të izoluara të lindjes janë jo vetëm të pamjaftueshme, por shpeshherë të gabuara.
Nevojat e praktikës klinike kontribuan në zgjerimin e punës kërkimore për të studiuar etiologjinë dhe patogjenezën e defekteve të shumta kongjenitale të zhvillimit. Ky seksion më vonë u quajt sindromologji. Një nga ilustrimet e qarta të ndryshimit midis sindromologjisë dhe mjekësisë klasike, d.m.th. mjekësia, ku diagnostikohet dhe studiohet një patologji e izoluar e një organi apo sistemi, është fakti se në mjekësia klasike vetëm disa sëmundje të reja janë përshkruar gjatë shekullit të 20-të ( sëmundje nga rrezatimi, Sëmundja e Legjionarëve, SIDA, sëmundja Lyme), ndërsa në sindromologji numri i formave të tilla nozologjike i kalonte 5000 dhe përshkruhen të paktën 80 të reja çdo vit.
Për disa forma të patologjisë sindromike, gjenetika molekulare moderne ka bërë të mundur lokalizimin e gjeneve që i përcaktojnë ato dhe studimin e produkteve të transkriptimit të gjeneve, të cilat më shpesh përfaqësohen nga receptorët e membranës ose faktorët e rritjes së indeve. Për shembull, në sëmundjen e Hirschsprung janë identifikuar dy mutacione të gjeneve të ndryshme: onkogjeni RET dhe receptori endotelin B, të cilët bënë të mundur dallimin e dy llojeve gjenetike të kësaj patologjie të lindur.
Sindromologjia është një fushë jashtëzakonisht e gjerë, që mbulon pothuajse të gjitha specialitetet e mjekësisë. Përafërsisht 1% e të gjithë të porsalindurve kanë shumëfish anomalitë kongjenitale ose sindromave. Prej tyre, në 40% të rasteve, një sindromë e caktuar tashmë mund të diagnostikohet sot, dhe 60% e mbetur kërkojnë ndarjen e tyre si sindroma "të reja". Megjithëse shumë sindroma janë mjaft të rralla, format sindromike të patologjisë në total përbëjnë një pjesë të rëndësishme sasiore të mjekësisë.
Tabela 36-10. Mekanizmat e përçarjes vaskulare në embrion dhe fetus (Cohen M.M., 1997)
| Faktorët e mjedisit | Efekt teratogjen |
| Preparate farmakologjike | |
| Talidomidi | Reduktimi i anomalive të ekstremiteteve. Hipoplazia e brezit të ekstremiteteve të sipërme. |
| Anomalitë e veshit | |
| Alkooli | Vonesa zhvillimin fizik. Fenotipi i pazakontë (qepalla të shkurtra) |
| lojëra elektronike). Mikrocefali, prapambetje mendore | |
| DietylstyleOestrol | Adenomatoza vaginale. Erozioni i qafës së mitrës. Adenokarcinoma vaginale |
| (rralle) | |
| warfarin | |
| Hipoplazia e kërceve të hundës. Defekte kongjenitale të sistemit nervor qendror. Kalcifikimi i pikës | |
| epifizat | |
| Hydantoin (dilantin) | |
| Zhvillimi fizik i vonuar. Fenotip i pazakontë. Mikrocefali, mendore | |
| prapambetja | |
| Trirmetadion | |
| Zhvillimi i vonuar psikomotor. Fenotip i pazakontë (i harkuar | |
| vetullat). Infeksioni i buzës ose qiellzës | |
| Aminopterina | |
| Metotreksat | Abortet spontane. hidrocefalus kongjenital. Zhvillimi fizik i vonuar |
| tiya. Fenotip i pazakontë | |
| Streptomicina | |
| Humbje kongjenitale e dëgjimit sensorineural | |
| Tetraciklina | |
| hipoplazi kongjenitale smalti i dhëmbëve. Ndryshim i ngjyrës së dhëmbëve (dhëmbët e verdhë) | |
| valproat natriumi | |
| Defektet e tubit nervor (Spina bifida) | |
| Retinol | |
| Abortet spontane. Anomalitë kraniofaciale. Defektet e tubit nervor | |
| Preparate litiumi | |
| Defektet kongjenitale të zemrës (anomalia Ebstein) | |
| Barnat antitiroide | |
| VG. Gusha | |
| Androgjene dhe doza të larta | |
| maskulinizues | Maskulinizimi |
| progestineve | |
| Penicilaminë | |
| Lëkurë hiperelastike. patologji kongjenitale IND lidhës | |
| Metilmerkur (merkur) | Substancat kimike |
| atrofi kongjenitale trurit. Spasticitet, konvulsione. mendore | |
| prapambetja | |
| Plumbi | |
| Zhvillimi fizik i vonuar. Ngjyrosje e pazakontë e lëkurës (gri) | |
| Pirja e duhanit | Ndikimet fizike, ushqyese dhe të tjera |
| Abortet spontane. IUGR | |
| rrezatimi jonizues | |
| Dëmi varet nga mosha e shtatzënisë. Abortet spontane. Kongjenitale | |
| defekte zhvillimore (dita 18-36 e shtatzënisë). Mikrocefalia dhe prapambetja mendore | |
| Humbur (java e 8-15 e shtatzënisë) | |
| hipertermia | |
| Defektet e lindjes të SNQ | |
| diabeti mellitus i nënës | |
| UPU. Sindroma e regresionit kaudal | |
| mungesa e jodit në ushqim | |
| PKU në nënë | Gusha. Prapambetja mendore dhe zhvillimi fizik i vonuar |
| Aborti, mikrocefalia, prapambetja mendore |
Nëse simptomat ose kompleksi i simptomave janë për shkak të një shkaku (etiologji monokauzale), atëherë termi sindrom nënkupton formën nozologjike të sëmundjes (sindromë nosologjike) dhe në këtë kuptim është sinonim me termin "sëmundje". Në praktikë, duke ndjekur rekomandimet e ekspertëve ndërkombëtarë, termi "sëmundje" përdoret më mirë në rastet me ecuri progresive të sëmundjes.
Kështu, sindroma është etiologjikisht sëmundje të caktuara me efekt pleiotropik (të shumëfishtë).
Një shembull i një sindrome të tillë është historia e izolimit të sëmundjes Recklinghausen, ose neurofibromatozës së tipit 1. Në 1849, Robert Smith, një kirurg kryesor në Shkollën Mjekësore të Dublinit, publikoi tiparet klinike dhe patoanatomike të dy rasteve të neurofibromatozës dhe citoi të dhëna nga 75 botime të mëparshme në literaturë mjekësore. Sidoqoftë, vetëm në mesazhin e Recklinghausen (von Recklinghausen, 1882) u vërtetua ideja e neurofibromatozës si një formë nozologjike. Tashmë është treguar se kjo patologji është një nga sëmundjet trashëgimore më të zakonshme të njeriut dhe shfaqet me një frekuencë prej 1 në 2000 lindje. Moderne kriteret diagnostike kjo sëmundje në bazë të tillë simptoma karakteristike të tilla si hiperpigmentimi i lëkurës (si "café au lait"), nyjet false kongjenitale ose lakimi i kockave të ekstremiteteve të poshtme, u zhvilluan vetëm në vitin 1987. Duhet theksuar se diagnoza është e mundur në rastet kur pacienti ka dy nga shenjat e mëposhtme, dhe me kusht që të mos jenë simptoma të ndonjë sëmundjeje tjetër.
Kriteret diagnostike për neurofibromatozën e tipit 1 (sëmundja Recklinghausen) (memorandumi i OBSH-së, 1992):
Gjatë ekzaminimit nën dritën artificiale të një pacienti që nuk ka arritur pubertetin, gjenden të paktën pesë pika pigmenti kafe të lehta (më shumë se 5 mm në pikën më të gjerë); kur ekzaminoni një pacient që ka arritur pubertetin - të paktën 6 njolla moshe (më shumë se 15 mm në pikën më të gjerë);
Prania, sipas anamnezës ose ekzaminimi klinik, dy ose më shumë neurofibroma të çdo lloji, ose një neurofibromë pleksiforme;
Të shumëfishta, të ngjashme me njollat njolla të errëta në sqetull ose ijë;
Displasia e krahut kocka sfenoidale ose lakimi kongjenital ose hollimi i kockave të gjata me formimin nyje e rreme ose pa të;
Glioma e nervit optik;
Dy ose më shumë njolla/nyje Pisch të zbuluara në iris gjatë ekzaminimit me llambë të çarë;
Prania e neurofibromatozës së tipit 1, sipas kritereve të mësipërme, në një të afërm të shkallës së parë (prind, vëlla apo vëlla apo pasardhës).
Diagnoza në kohë e neurofibromatozës Recklinghausen kërkon vëzhgim dinamik me CT periodike të kokës dhe palca kurrizore me qëllim të diagnoza e hershme neoplazia e SNQ.
Tashmë në vitet 60-70 të shekullit XX. janë përshkruar shumica e sindromave kromozomale dhe teratogjene dhe sasi e madhe sindromat e gjeneve. Nga fillimi i viteve 80 të shekullit XX. Përvoja lejoi jo vetëm zhvillimin e një terminologjie të unifikuar ndërkombëtare, por edhe të propozonte disa qasje metodologjike për identifikimin e sindromave "të reja" dhe studimin e patogjenezës së defekteve të shumta zhvillimore (Spranger J. et al, 1982; Cohen MM., 1982; Cohen MM. , 1997).
Në praktikë, format sindromike të patologjisë përfshijnë edhe rastet kur, përveç një defekti të vetëm të lindur, tek një fëmijë vërehet një fenotip i pazakontë, domethënë prania e tre ose më shumë anomalive të vogla zhvillimore.
Anomalitë e vogla zhvillimore ose stigmat e disembriogjenezës janë variante morfologjike anormale organet individuale ose pëlhura që nuk kanë vlera mjekësore, d.m.th. që nuk kërkon trajtim. Shfaqja e këtyre varianteve shoqërohet me embrionale ose, më rrallë, me periudha pjellore morfogjeneza e njeriut. NË gjenetike klinike dhe sindromologjia, anomalitë e vogla zhvillimore janë një shenjë diagnostike jashtëzakonisht e rëndësishme, që tregon një probabilitet të lartë shkelje të rënda morfogjeneza në formën e keqformimeve kongjenitale që kërkojnë diagnostifikimi i veçantë dhe shpesh ndërhyrje kirurgjikale të mëvonshme (Tabela 36-11).
Më shumë se 200 variante morfogjenetike informative janë përshkruar te njerëzit, megjithëse praktika klinike zakonisht nuk përdoren më shumë se 80 anomali të vogla zhvillimore.
Anomali të vogla zhvillimore tek të porsalindurit:
Kreu:
V model jonormal i rritjes së flokëve;
V zverku i rrafshuar;
V "gunga" e kasafortës kraniale.
Zona orbitale:
V palosje epikantike;
V epikant i kundërt;
V Seksioni mongoloid i syve;
V seksion anti-mongoloid i syve;
V çarje të shkurtra palpebrale;
V dystopia e qosheve të jashtme të syrit;
V hipotelorizëm i moderuar;
V hipertelorizëm i moderuar;
Ptoza është e lehtë;
V heterokromi e irisave;
V mikrokornea.
Predhat e veshit:
V forma primitive;
V tuberkulozi i Darvinit;
V formë jonormale e kaçurrelave;
V aurikulat asimetrike;
V aurikulat e rrotulluara;
V aurikulat e reduktuara;
V aurikulat e dala;
V mungesa e një tragus;
Ndarja e urinës;
V mungesa e një lobi;
V gropa veshore;
V "daljet" e veshit;
V ngushtimi i kanalit të jashtëm të dëgjimit.
Hunda dhe filtri:
¦¦¦ hipoplazia e krahëve të hundës;
¦¦¦ vrimat e hundës të vendosura;
¦¦¦ filtër i rrafshuar;
¦¦¦ filtri i dalë.
zonën e gojës dhe zgavrën e gojës:
¦¦¦ mikrogjenia;
¦¦¦ çarje e gjuhës;
¦¦¦ frenulum aberrant i vestibulës së gojës;
¦¦¦ dhëmbë-filtër neonatal.
¦¦¦ qafa pterygoid - mesatarisht;
¦¦¦ fistula të qafës.
Polidaktilia rudimentare; e vetmja palosje e përkuljes së pëllëmbës; dermatoglife jonormale; klinodaktilia e gishtave të vegjël; shkurtimi i gishtave 4-5; falangat terminale hipoplastike.
sindaktili P-Sh gishtat; boshllëqe sandale; gishti i parë i shkurtër; imponimi i gishtave (IV-V); thonjtë e trashë.
Mbulesa të lëkurës:
Hemangioma;
Hiperpigmentimi i lëkurës dhe nevi; Njolla mongoloide (në racën e bardhë); depigmentimi i lëkurës; thithkat apo areolat aksesore.
Trupi:
¦¦¦ diastaza e muskujve rectus abdominis;
¦¦¦ hipospadia e moderuar (kokat);
¦¦¦ mbresa të thella të sakrumit.
Skeleti:
¦¦¦ depresioni ose zgjatja e sternumit.
Koka, qafa dhe dora janë më informueset në lidhje me këto shenja, më shumë se 70% e të gjitha anomalive të vogla zhvillimore ndodhen në këtë zonë. Vlera diagnostike anomalitë e vogla zhvillimore janë të ndryshme. Është thelbësisht e rëndësishme që rëndësia praktike e këtyre shenjave qëndron në diagnostikimin e tre ose më shumë anomalive. Çdo i porsalindur me tre ose më shumë anomali të vogla zhvillimore ka një probabilitet të lartë (90%) për një keqformim serioz kongjenital të trurit, zemrës, veshkave ose shtyllës kurrizore, përveç kësaj ka probabilitet të lartë(50%) diagnoza e formës sindromike të patologjisë. Nëse një fëmijë me një vonesë në shkallën e zhvillimit psikomotor ka tre ose më shumë anomali të vogla zhvillimore, Rreziku i lartë dëmtimi organik CNS (shih Tabelat 36-11). Ndonjëherë vetëm prania e dy anomalive të vogla zhvillimore është informative për diagnozën. Për shembull, hipotelorizmi (i ndarë ngushtë kokërdhokët e syve) dhe e vetmja prerësi i sipërm tregojnë një keqformim kongjenital të trurit siç është grupi prosencefalik.
Diagnoza e një defekti kongjenital të zhvillimit tek një fëmijë shtron pyetjet e mëposhtme për neonatologun:
Cilës lloj patologjie i përket ky defekt (keqformim kongjenital, përçarje, deformim apo displazi)?
Sa shpesh ka lidhje me këtë defekt të lindjes në formën e defekteve të tjera të lindjes ose sëmundjeve që ende nuk janë manifestuar klinikisht?
Me çfarë shpeshtësie është ky defekt kongjenital një simptomë e formës sindromike të patologjisë?
Cilat sindroma janë më të shpeshta në këtë defekt kongjenital?
Përgjigjet për këto pyetje janë të parat faza diagnostike praktike punë e përbashkët neonatolog dhe gjenetist. Objektivi final kjo fazë është diagnoza e defekteve kongjenitale shtesë të zhvillimit ose diagnoza e një sindromi specifik. Në rastin e diagnostikimit të një forme sindromike të patologjisë, në shumicën e rasteve bëhet e qartë më tej taktika mjekësore për konservatore ose trajtim kirurgjik dhe prognoza gjenetike mjekësore në familjen e një fëmije të sëmurë. Informacion rreth prognozës për jetën dhe shëndetin në një sindromë të veçantë ka rëndësi të madhe dhe është qëllimi kryesor i punës mjekësore.
Sindromat mund të analizohen në nivele të ndryshme të organizimit biologjik:
Në nivel të çrregullimeve brenda proceseve metabolike (sindromat dismetabolike);
Në nivel të çrregullimeve të indeve (sindromat e displazisë);
Në nivelin e çrregullimeve të morfologjisë së organeve (sindromat e keqformimeve kongjenitale dhe sindromat e disruptimit);
Në nivelin e shkeljeve të një pjese të caktuar të trupit (sindromat e deformimeve). Të katër nivelet e shkeljeve kanë dhe vlerë praktike, meqenëse të katër llojet biologjike të sindromave kanë kritere të qarta diagnostike dhe gjenetike për diagnostikimin dhe prognozën për shëndetin (Tabela 36-12).
Diagnoza e formave sindromike të patologjisë tek një i porsalindur përfshin:
Mundësi diagnozë të saktë sëmundjet shoqëruese një fëmijë me defekte të shumta;
Prognoza e komplikimeve karakteristike për çdo sindromë gjatë operacionit ose trajtim konservativ defekte të lindjes ose sëmundje;
Një vlerësim i saktë i mundësive të trajtimit kirurgjik ose konservativ të sëmundjeve (koha dhe shtrirja e ndërhyrjes kirurgjikale, rezultatet afatgjata trajtim);
Prognoza e saktë gjenetike mjekësore në familje.
Këto gjetje ilustrohen mirë nga vëzhgimet e mëposhtme nga praktika klinike.
Tabela 36-12. llojet biologjike sindromat (Cohen M.M., 1982)
| Lloji i sindromës (niveli i çrregullimeve) | Veçoritë karakteristike | Shembuj |
| Sindroma dismetabolike (metabolizmi) Sindroma e displazisë (indeve) | Të porsalindurit kanë një fenotip normal me një klinikë progradient. Shenjat klinike relativisht të ngjashme në krahasim me llojet e tjera të sindromave. Nuk ka lidhje me keqformime kongjenitale. Verifikimi i defektit primar biokimik është i mundur. Mënyra autosomale recesive e trashëgimisë Sindroma e displazisë së thjeshtë karakterizohet vetëm nga lezione | PKU, sëmundja Tay-Sachs, sindroma HCV Marfan, sindroma Ehlers-Danlos, akondroplazia |
| një shtresë embrionale. | ||
| Lloji dominant ose recesiv | ||
| trashëgimisë | ||
| Hamartoneoplastike | Neurofibromatoza | |
| sindromi: | Dy ose tre qeliza germinale janë të përfshira | Recklinghausen |
| fletë; | ||
| tendenca për neoplazi; | ||
| zakonisht mënyra dominuese e trashëgimisë | ||
| Sindromi i keqformimeve ose | sindromi down, | |
| ndërprerje (organet) | Dy ose më shumë defekte zhvillimore në një | sindromi TAR, |
| i porsalindur. Karakterizuar | sindromi i alkoolit | |
| pleiotropia embrionale. | fetusit | |
| Verifikimi biokimik | ||
| e pamundur ose e rrallë. Etiologjia | ||
| të ndryshme - monogjenike, kromozomale | ||
| ose teratogjene | ||
| Sindroma e deformimit (zona | ||
| trup) | Shkelje e formës ose e pozicionit | Sindroma Potter |
| formuar fillimisht normalisht | ||
| organeve apo pjesëve të trupit. | ||
| Shumica e rasteve shpjegohen | ||
| shkelje aktiviteti motorik | ||
| fetusi (hipokinezi). | ||
| Sistemi musculoskeletal zakonisht preket | ||
| sistemi. |
Një defekt i lindjes është një anomali në strukturën e trupit ose kiminë e një foshnjeje të porsalindur. Kjo mund të shkaktohet faktorët trashëgues (arsye gjenetike), si rezultat i ndikimit mjedisi që prek embrionin ose fetusin në mitër, ose një kombinim faktorësh. Shpesh, shkaqet e defekteve të lindjes nuk mund të përcaktohen.
Defektet e lindjes nganjëherë quhen anomali kongjenitale. Anomalitë që janë të pranishme në lindje përgjithësisht nuk konsiderohen si defekt i lindjes, përveç nëse ato rezultojnë në sëmundje, paaftësi fizike ose mendore. Për shembull, nishanet rrallë konsiderohen si një defekt në lindje, sepse zakonisht nuk shkaktojnë probleme shëndetësore.
Është vlerësuar se midis 3 dhe 5 për qind e fëmijëve kanë një lloj defekti të lindjes. Disa defekte të lindjes ndodhin rrallë. Të tjera, të tilla si disa defekte të lindura të zemrës, janë më të zakonshme.
Faktorët trashëgues dhe defektet e lindjes
Secili prej nesh ka gjene të trashëguara nga prindërit. Gjenet përcaktojnë karakteristikat ose tiparet tona të lindura. Në rastin e defekteve të lindjes, gjenet gjithashtu mund të ndikojnë në anomalitë.
Një fëmijë trashëgon dy kopje të secilit gjen, një nga nëna dhe një nga babai. Nëse gjeni me defekt është dominant, fëmija që trashëgon kopjen e dhënë do ta ketë defektin. Kjo për shkak se kopja e dëmtuar "dominon" mbi kopjen normale të trashëguar nga prindi tjetër. Por nëse gjeni i dëmtuar është recesiv, fëmija do të trashëgojë dy kopje të dëmtuara, një nga nëna dhe një nga babai.
Shembuj të defekteve të lindjes autosomale dominante janë sëmundja e Huntingtonit, një çrregullim i sistemit nervor, sindroma e Marfanit, e cila karakterizohet nga kocka të zgjatura dhe probleme të zemrës. Disa sëmundje kongjenitale, si sëmundja e Huntingtonit, nuk kanë simptoma për shumë vite.
Defektet e tjera të lindjes përcaktohen nga gjenet në kromozomin X (kromozomet X dhe Y përcaktojnë seksin e foshnjës). Hemofilia, çrregullimet kongjenitale të gjakut dhe verbëria e ngjyrave janë shembuj të defekteve të lindjes të lidhura me X.
Megjithatë, shumë defekte trashëgimore të lindjes nuk janë thjesht dominante, recesive ose të lidhura me X; ndodhin edhe gjene të shumta defektive.
Anomalitë kromozomale
Disa defekte të lindjes shkaktohen nga kromozome shtesë, të munguara, të paplota ose të keqformuara. Sindroma Down, një nga defektet më të zakonshme të lindjes, zakonisht shkaktohet nga prania e një kromozomi shtesë në qeliza. Sindroma Down karakterizohet me vonesë mendore, shtat të shkurtër dhe shenjat dalluese fytyrat. Defektet që lidhen me kromozomet seksuale mund të çojnë në probleme në zhvillimin seksual, duke përfshirë sterilitetin (pamundësia për të pasur fëmijë).
Faktorët e mjedisit
Defektet e lindjes mund të shkaktohen edhe nga faktorë mjedisorë. Mjedisi këtu i referohet trupit të nënës, megjithatë, shkencëtarët po studiojnë ndikimi i mundshëm mbi shfaqjen e defekteve të lindjes dhe kushteve mjedisore të Tokës.
 Gratë shtatzëna që konsumojnë shumë fazat e hershme shtatzënia kanë rrezik i rritur lindin fëmijë me fetale sindromi i alkoolit. fëmijët me të sëmundje kongjenitale mund të ketë defekte të ndryshme në rritje, pamje dhe aftësitë mendore. Dhe edhe konsumimi i moderuar i alkoolit mund të dëmtojë fetusin. Pirja e duhanit gjatë shtatzënisë bën që foshnja të ketë një peshë më të ulët të lindjes dhe rrit rrezikun e defekteve të tjera të lindjes.
Gratë shtatzëna që konsumojnë shumë fazat e hershme shtatzënia kanë rrezik i rritur lindin fëmijë me fetale sindromi i alkoolit. fëmijët me të sëmundje kongjenitale mund të ketë defekte të ndryshme në rritje, pamje dhe aftësitë mendore. Dhe edhe konsumimi i moderuar i alkoolit mund të dëmtojë fetusin. Pirja e duhanit gjatë shtatzënisë bën që foshnja të ketë një peshë më të ulët të lindjes dhe rrit rrezikun e defekteve të tjera të lindjes.
Disa sëmundje te gratë shtatzëna, për shembull, mund të çojnë në shurdhim, verbëri dhe defekte të lindura të zemrës tek foshnja. Sëmundjet veneriane mund të kalojë tek fetusi ose tek i porsalinduri.
Disa medikamente shoqërohen me defekte të lindjes. Më i njohuri është talidomidi, qetësues. Shumë barna të tjera, duke përfshirë qetësuesit, antibakterialët dhe barna antikancerogjene mund të shkaktojë anomali kongjenitale.
Të tjera faktorët e mjedisit që mendohet se rrisin rrezikun e defekteve të lindjes përfshijnë të ushqyerit e dobët dhe mosha e nënës. Për shembull, sa më e vjetër të jetë një grua shtatzënë, aq më shumë ka gjasa që ajo të lindë një fëmijë me sindromën Down.
Diagnoza e defekteve të lindjes
 Disa defekte të lindjes mund të diagnostikohen ndërsa foshnja është ende në mitër. Procedura që përdor valët ultrasonike për të marrë një imazh të fetusit në ekran, mund të ndihmojë mjekun të zbulojë disa keqformime. Për shembull, disa defekte të shtyllës kurrizore mund të zbulohen duke përdorur ultratinguj.
Disa defekte të lindjes mund të diagnostikohen ndërsa foshnja është ende në mitër. Procedura që përdor valët ultrasonike për të marrë një imazh të fetusit në ekran, mund të ndihmojë mjekun të zbulojë disa keqformime. Për shembull, disa defekte të shtyllës kurrizore mund të zbulohen duke përdorur ultratinguj.
Në një procedurë të quajtur , ekzaminohet një mostër e vogël e lëngut që rrethon fetusin. Ky test është i dobishëm për zbulimin e defekteve të lindura metabolike dhe anomalive kromozomale.
Shumë defekte të lindjes mund të diagnostikohen nga ekzaminimi fizik i një foshnjeje të porsalindur nga mjeku. Teste të tjera duke përfshirë rrezet X mund të përdoret nëse mjekët dyshojnë për defekte të lindjes. Testet e gjakut mund të zbulojnë anomali të caktuara në gjak ose në kiminë e trupit.
Trajtimi i defekteve të lindjes
Jo çdo defekt në lindje ndikon në cilësinë e jetës së personit që e ka atë. Disa defekte të lindjes kanë pak efekt, përveç ndoshta në pamje.
Disa defekte të lindjes mund të trajtohen për t'i parandaluar ose zvogëluar ato. efekt i dëmshëm. Kirurgjia mund të kryejë operacione për të korrigjuar keqformimet kongjenitale të tilla si shputa e shputës, qiellza e çarë, buzë e çarë, strukturore dhe traktit gastrointestinal. Trajtimi mund të zvogëlojë simptomat e fibrozës cistike, sëmundje trashëgimore të lidhura me frymëmarrjen. Në disa raste, çrregullime të tilla si hidrocefalusi mund të korrigjohen para lindjes.
 Mjekët ndonjëherë mund të trajtojnë çrregullime kongjenitale kimia e trupit përmes barnave dhe dieta speciale. Për shembull, trajtim të shpejtë mund të parandalojë dëmtimin e trurit te fenilketonuria, një defekt metabolik që mund të çojë në të rënda prapambetje mendore. Edukim special, rehabilitimi, si dhe përdorimi i pajisjeve dhe pajisjeve speciale mund të ndihmojë në kompensimin e disa prej mendore dhe të meta fizike të tilla si verbëria dhe shurdhimi.
Mjekët ndonjëherë mund të trajtojnë çrregullime kongjenitale kimia e trupit përmes barnave dhe dieta speciale. Për shembull, trajtim të shpejtë mund të parandalojë dëmtimin e trurit te fenilketonuria, një defekt metabolik që mund të çojë në të rënda prapambetje mendore. Edukim special, rehabilitimi, si dhe përdorimi i pajisjeve dhe pajisjeve speciale mund të ndihmojë në kompensimin e disa prej mendore dhe të meta fizike të tilla si verbëria dhe shurdhimi.
Parandalimi i defekteve të lindjes
Askush nuk mund të garantojë që një fëmijë do të lindë “perfekt” dhe i shëndetshëm. Megjithatë, ka mënyra për të minimizuar mundësinë që një fëmijë të ketë defekte të lindjes.
Gratë shtatzëna nuk duhet të pinë duhan ose të përdorin pije alkolike ata nuk duhet të marrin ilaçe të çfarëdo lloji, përveç rasteve kur ju rekomandon mjeku. Disa vitamina, kur merren në sasinë e duhur, mund të ndihmojnë në parandalimin e disa defekteve të lindjes. Për shembull, acid folik gjatë shtatzënisë mund të ndihmojë në parandalimin e disa defekteve të shtyllës kurrizore dhe të sistemit nervor qendror. Vaksinimi shumë përpara shtatzënisë mund të parandalojë defektet e lindjes që mund të vijnë nga sëmundjet gjatë shtatzënisë.
Mohimi i përgjegjësisë: Informacioni i dhënë në këtë artikull mbi defektet e lindjes ka për qëllim të informojë vetëm lexuesin. Nuk mund të zëvendësojë këshillën e një profesionisti shëndetësor.
