Системна амилоидоза: диагностика, диференциална диагноза, лечение. Клинично протичане и основните му прояви
Амилоидоза- заболяване, характеризиращо се с метаболитно нарушение, в резултат на което се образува ново за организма вещество (амилоид), което се отлага в органите и нарушава техните функции.
Амилоидът е сложен гликопротеин, в който фибриларните и глобуларните протеини са тясно свързани с полизахаридите. Амилоидният фибрил се състои от полипептидни протеини; Освен фибриларен протеин, амилоидът съдържа и друг протеин - така нареченият Р-компонент, който е еднакъв във всички форми на амилоид. Смята се, че P компонентът е нормален серумен протеин, свързан с амилоидни фибрили.
Амилоидозата може да възникне като усложнение на всяко заболяване или да се развие като независим процес.
В момента, в зависимост от етиологията, се разграничават няколко форми на амилоидоза, които имат свой собствен биохимичен състав на амилоидни фибрили.
Първичната (идиопатична) амилоидоза се развива без видими причинии засяга различни органи (сърце, бъбреци, черва, черен дроб, нервна система). Биохимичната форма на първичната амилоидоза е AL формата; предшественикът на такъв амилоид е Ig и леките вериги на имуноглобулините. Според структурата на амилоида и естеството на лезията вътрешни органиАмилоидозата при мултиплен миелом, която в момента се класифицира като отделна група, е близка до първичната (идиопатична) доза амилоид.
Наследствената (генетична) амилоидоза се проявява с преобладаващо бъбречно увреждане, комбинация от бъбречно увреждане и нервна система. У нас наследствената амилоидоза обикновено се свързва с периодично заболяване, което се предава по автозомно-доминантен път. При това заболяване амилоидозата може да бъде единствената проява. Биохимичната форма на наследствената амилоидоза е ПМ (предшественикът на амилоида е преалбумин). При периодичното заболяване биохимичната форма е АА (предшественикът е SAA протеинът).
Придобитата (вторична) амилоидоза се среща най-често и се развива при ревматоиден артрит, анкилозиращ спондилит, туберкулоза, хронично нагнояване - остеомиелит, бронхиектазии, хроничен белодробен абсцес, по-рядко - при неспецифичен улцерозен колит, псориазис, лимфогрануломатоза, сифилис, тумори на бъбреците и белите дробове и др. Биохимичната форма на вторична амилоидоза е АА (нейният серумен предшественик е SAA протеинът, синтезиран от хепатоцитите).
Сенилната амилоидоза е резултат от инволютивни нарушения на протеиновия метаболизъм, открити в мозъка, панкреаса и сърцето. Биохимична формула - AS (прекурсорът е преалбумин).
Локалната амилоидоза се развива без видима причина, нейната биохимична формула е AE (прекурсор неизвестен).
Патогенеза.Добре известни са само отделни връзки на патогенезата (Схема 22). От диаграмата следва, че под влияние на генната мутация, както и влиянието външни факториимунитетът се променя - намалява
брой Т-лимфоцити. Това води до намаляване на контролиращия им ефект върху В-лимфоцитната система. В резултат на това броят на В-клетките, носещи нормални имуноглобулинии броят на В клетките, които синтезират прекурсори на амилоидни фибрили, се увеличава. Амилоидобластите произвеждат фибриларен протеин в повишени количества, което предизвиква синтеза на амилоид в големи количества.
Въпреки това, поради генетичен дефект в амилоидокластите, който допринася за намаляване на тяхната ензимна активност, не настъпва достатъчна резорбция на амилоид. В резултат на това се наблюдава повишено отлагане на амилоид в тъканите и органите [Mukhin N.A., 1981].
При мултиплен миелом амилоидозата се развива в резултат на повишено производство на парапротеин от плазмените клетки, което върви към изграждането на амилоид. Съставът на амилоида при различните форми на амилоидоза е различен, което се определя от състава на протеина на амилоидните фибрили.
С увреждане на миокарда, периферни нерви(наблюдава се главно при идиопатична амилоидоза) амилоидът се отлага около колагенови влакна съединителната тъкан. При лезии се наблюдава отлагане на амилоид около ретикуларните влакна
бъбреци, черва, черен дроб, надбъбречни жлези, панкреас (с наследствена и вторична амилоидоза). Въпреки това е възможна комбинация от периколагенно и периретикуларно отлагане на амилоид, което осигурява комбинирани лезии на различни органи и системи.
Когато амилоидът се отложи в тъканите, броят на функциониращите елементи, кардиомиоцитите, хепатоцитите, нервните влакна и бъбречните гломерули намалява, което впоследствие води до развитие на органна недостатъчност.
Така в сърцето амилоидът се отлага под ендокарда, в стромата и съдовете на миокарда, както и по протежение на вените в епикарда. В същото време сърцето рязко се увеличава по размер и броят на кардиомиоцитите бързо намалява. Всичко това води до намаляване контрактилна функциямиокардна и сърдечна недостатъчност, както и нарушения в проводимостта и сърдечния ритъм. В мозъка със сенилна амилоидоза амилоидът се намира в така наречените сенилни плаки на кората, съдовете и мембраните. В кожата амилоидът се отлага в папилите и стените на кръвоносните съдове, което води до тежка атрофия на епидермиса. В черния дроб амилоидът се отлага между стелатните ретикулоендотелиоцити на синусоидалните съдове, в стените на кръвоносните съдове, каналите и в съединителната тъкан на порталните пътища. Тъй като амилоидът се натрупва, чернодробните клетки атрофират.
В бъбреците амилоидът се отлага в мембраната на гломерулните капиляри и тубулите на нефрона, в мезангиума, капилярните бримки и по дължината на артериолите. Тъй като амилоидът се натрупва, повечето нефрони атрофират, умират или се заместват от съединителна тъкан - появява се амилоидно набръчкан бъбрек. Този процес може да бъде представен като следната диаграма:
протеинурия -> нефротичен синдром -> бъбречна недостатъчност.
Съответно в клиничната картина се разграничават три етапа: 1) начален (протеинурен); 2) разширен (нефротичен); 3) терминал (азотемичен).
Клинична картина.Проявите на амилоидоза са разнообразни и се определят от: 1) локализация на амилоид в определен орган; 2) тежестта на амилоидните отлагания в органа; 3) основното заболяване, срещу което се е развил амилоид (във вторичната форма на амилоидоза).
По време на диагностицирането могат да възникнат трудности поради факта, че клиничните прояви на заболяването ще бъдат забележими само при определено количество отложен амилоид. В тази връзка е неизбежен "латентен" период от момента на отлагане на амилоид до появата на симптоми на дисфункция на орган или система.
Клиничната картина е особено впечатляваща в случаите на увреждане на бъбреците, най-честата локализация на амилоидните отлагания.
На етап I диагностично търсене V начална фазаНе може да се получи почти никаква информация, показваща увреждане на бъбреците от амилоидоза. Оплакванията на пациентите са свързани с основното заболяване (вторична амилоидоза).
Анамнезата съдържа информация за наличието на дадено заболяване (белодробна туберкулоза, остеомиелит, ревматоиден артрит и др.), неговия ход и проведената терапия. Тази информация сама по себе си не позволява диагностицирането на бъбречна амилоидоза, но предупреждава лекаря за тази възможност.
В напреднал стадий на амилоидоза пациентите се оплакват от развитието на нефротичен синдром, намаляване на количеството на урината, оток с различно разпространение и тежест, както и оплаквания от слабост, липса на апетит и намалена работоспособност. Наред с тях, при вторична амилоидоза има оплаквания от проявата на основното заболяване.
В терминалния стадий оплакванията са причинени от развиваща се хронична бъбречна недостатъчност: загуба на апетит, гадене, повръщане (диспептични разстройства), главоболие, нарушения на съня (заболявания на нервната система), сърбеж.
На етап II от диагностичното търсене могат да бъдат открити на ранен етап само симптоми, характерни за основното заболяване (при вторична амилоидоза).
IN напреднал стадийоткриване: 1) оток с различна локализация и тежест; при значително задържане на течности в тялото може да се появи хидроторакс, хидроперикард и преходен асцит; 2) артериална хипертония (среща се при 12 - 20% от пациентите с амилоидоза), дилатация и хипертрофия на лявата камера; 3) уголемяване на черния дроб и далака поради отлагане на амилоид в тъканите (черният дроб и далакът са плътни, безболезнени, със заострен ръб); 4) симптоми на основното заболяване (с вторична амилоидоза).
IN терминален стадийсимптомите се определят от тежестта на бъбречната недостатъчност: 1) дистрофичен синдром (промени в кожата и лигавиците); 2) серозно-ставен синдром (остеоартропатия, вторична подагра, сух перикардит, плеврит); 3) артериална хипертония.
На етап III от диагностичното търсене на амилоидоза се получава най-значимата информация за поставяне на диагнозата, която може да бъде групирана по следния начин: 1) уринарен синдром; 2) нарушения на протеиновия и липидния метаболизъм; 3) откриване на отлагания на амилоидни маси.
Уринарен синдром: 1) протеинурия - най-важният симптомамилоидозата се развива във всичките й форми, но най-често при вторична амилоидоза. Протеинурията обикновено е значителна, на ден се отделят 2-20 g протеин, основната част от които е албумин. Глобулините се освобождават в по-малки количества и серумният амилоиден прекурсор (SAA протеин) може да се екскретира в урината. В терминалния стадий протеинурията продължава. В урината могат да бъдат открити a- и особено y-гликопротеини.
Според степента на протеинурия се откриват хиалинни и по-рядко гранулирани отливки. Микрохематурия или левкоцитурия рядко се диагностицира, но нейната тежест не съответства на степента на протеинурия (както се наблюдава при гломерулонефрит). Степента на нарушения на липидния метаболизъм при амилоидоза съответства на липоидурията с наличието на двойнопречупващи кристали в седимента на урината.
Нарушения на протеиновия и липидния метаболизъм: 1) хипопротеинемия в комбинация с хипоалбуминемия и хипер-<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
От голямо значение за диагнозата откриване на амилоидни маси в органи и тъкани:в черния дроб (50% от случаите), далака (с пункционна биопсия), лигавицата на венците и ректума.
В началния етап (протеинуричен) биопсията на лигавицата на венците често дава отрицателен резултат, а ректума - положителен резултат; в напреднал стадий (нефротичен) в първия случай резултатите са положителни при половината от пациентите, а във втория - още по-често.
И накрая, при хронична бъбречна недостатъчност данните от биопсията на тъканта на венците са положителни в повече от половината от случаите, а на ректалната лигавица в почти всички случаи. Затова при напреднал процес трябва да се препоръча биопсия на гингивалната лигавица, а при всеки стадий на амилоидозата – ректална биопсия.
Ако се подозира идиопатична амилоидоза (проявява се по-често с увреждане на сърцето, периферните нерви, по-рядко - бъбреците), препоръчително е преди всичко да се направи биопсия на лигавицата на венците, а в случай на вторична (придобита) амилоидоза и нейната наследствени форми (възникват с преобладаващо увреждане на бъбреците) - биопсия на лигавицата на ректума.
Редица други изследвания помагат: 1) да се изясни диагнозата на заболяването, на фона на което се е развила амилоидозата; 2) оценка на функционалното състояние на бъбреците (тест на Rehberg, Zimnitsky, ниво на креатинин в кръвта).
Поток. Клиничната картина на бъбречната амилоидоза има характеристики, които я отличават от бъбречно увреждане с различен произход: 1) нефротичният синдром се развива постепенно и често след дълъг стадий на протеинурия, характеризира се с персистиращ курс, отокът често е резистентен към различни диуретици. При CGN нефротичният синдром обикновено се появява в началото на заболяването и често се повтаря в бъдеще; 2) артериалната хипертония се наблюдава рядко, дори в стадия на хронична бъбречна недостатъчност; 3) при първична амилоидоза хроничната бъбречна недостатъчност е по-доброкачествена за разлика от вторичната амилоидоза или CGN (поради по-малката тежест на гломерулното увреждане в сравнение с вторичните форми на амилоидоза); 4) ходът на вторичната амилоидоза до голяма степен зависи от основното заболяване, при чести екзацербации на което е възможно значително прогресиране на амилоидозата.
Усложнения. При амилоидоза в 2-5% от случаите се развиват:
1) тромбоза на бъбречната вена (с вторична амилоидоза), която се проявява
хематурия и болка в лумбалната област, повишаване на протеина
риа и спад в диурезата;
2) интеркурентна инфекция;
3) фибринозно-гноен перитонит, чиято поява е придружена
има рязко увеличаване на асцита.
Диагностика.Клиничните прояви на амилоидозата са неспецифични. Всеки от симптомите (оток, протеинурия, артериална хипертония) може да се появи при различни бъбречни заболявания. Единственият метод за надеждно диагностициране на амилоидозата е органна биопсия (бъбреци, черен дроб, ректална лигавица или дъвка), но тя не винаги е осъществима. Ето защо в повечето случаи е необходимо да се съсредоточи върху клиничните прояви на патологичния процес.
Наличието на заболяване, при което вторичен
амилоидоза (клинични или анамнестични признаци).
Началото и прогресирането на протеинурия или началото
нефротичен синдром.
Няма заболяване, при което да се развие амилоидоза,
въпреки това има протеинурия или нефротичен синдром.
Наличието на персистираща тежка сърдечна недостатъчност, синдромът не е такъв
недостатъчност на абсорбция, полиневропатия (ако в същото време последният
Трудно е да се обяснят трите синдрома с други причини).
Наличието на амилоидоза може да се предположи със следните лабораторни признаци на нефротичен синдром (който, както е известно, може да се развие при други бъбречни заболявания):
а) тежка диспротеинемия + хипоалбуминемия + хипер-sg- и gi-
пергамаглобулинемия;
б) повишени нива на аг-гликопротеин, р-липопротеини;
в) появата в урината на а- и особено у-гликопротеини и а-липопротеини
Тейдов.
Във всички случаи вероятността от развитие на амилоидоза се увеличава с откриването на хепато- и спленомегалия, както и сърдечни промени, характерни за амилоидоза (в такива случаи говорим за идиопатична генерализирана амилоидоза).
Следователно диагнозата бъбречна амилоидоза може да се постави с достатъчна увереност в напреднал (нефротичен) или терминален стадий, докато в началния (протеинурен) стадий това е много по-трудно да се направи. В тези случаи преходната или постоянна протеинурия трябва да се диференцира от гломерулонефрит (остър, хроничен). Трябва да се вземе предвид следното:
1) по-бавно прогресиране на бъбречно увреждане с амилоид
доза;
2) липсата на ясна връзка при амилоидоза с настинки
ниями;
3) постоянното наличие на микрохематурия при гломерулонефрит (с
амилоидоза в 20% от случаите).
Понякога правилната диагноза може да се постави само след период на дългосрочно наблюдение на пациента. Въпросът се решава много по-бързо, ако е възможно да се извърши пункционна биопсия на бъбрека.
Формулиране на подробна клинична диагнозаамилоидоза взема предвид следните компоненти: 1) формата на амилоидоза; 2) стадий на амилоидоза (протеинурен, нефротичен, терминален); 3) функционално състояние на бъбреците (липса или наличие на бъбречна недостатъчност, степента на нейната тежест); 4) основно заболяване (с вторична амилоидоза); 5) състоянието на други органи (сърце, черен дроб, нервна система и др.) С идиопатична (първична) амилоидоза.
Лечение.Проблемът с лечението на амилоидозата все още остава нерешен, тъй като причините, водещи до повишена амилоидогенеза и недостатъчна резорбция, не са ясни. Въпреки това е възможно да се проведат редица терапевтични мерки, които подобряват състоянието на пациента. Понастоящем лечението на пациент с амилоидоза се извършва, като се вземат предвид: 1) въздействието върху основното заболяване, срещу което се е развила амилоидозата
(втори); 2) влияние върху механизмите на патогенезата; 3) въздействие върху основните клинични синдроми.
Въздействие върху основното заболяване, срещу което се развива
вторична амилоидоза се налага поради факта, че чести
напрежение или висока активност на патологичния процес водят до
прогресия на амилоидозата.
Това въздействие е както следва:
а) при хронични инфекции (туберкулоза, сифилис) е необходимо
дългосрочна специфична терапия;
б) за хронични неспецифични белодробни заболявания - ком
комплексна терапия с използване на антибиотици, бронхиален дренаж и
ако е необходимо, хирургическа интервенция (например при хронични
логичен белодробен абсцес);
в) със системни заболявания на съединителната тъкан, например с
ревматоиден артрит, комплексна терапия е показана, включително
значението на основните лекарства (D-пенициламин, златни соли, аминокиселини
нолинови лекарства).
Въздействието върху механизмите на патогенезата включва намаляване
амилоиден синтез:
а) дневен прием на 80-120 g суров черен дроб в продължение на 6-12 месеца
води до намаляване на протеинурията, намаляване на размера на черния дроб
нито далака;
б) аминохинолинови лекарства (хингамин или делагил, според
0,25 - 0,5 g на ден в продължение на много месеци и дори години) намалява
има прогресия на процеса. Очевидно тези средства за въздействие
участват в синтеза на амилоидни фибрили. Лечението е ефективно само
в ранните стадии на амилоидоза; когато процесът е много напреднал
(пълен нефротичен синдром, бъбречна недостатъчност
ity) предписването на тези лекарства е неподходящо;
в) с развитието на амилоидоза поради периодично повтарящо се заболяване
Препоръчва се колхицин;
г) при първична амилоидоза се предписва и мелфалан, който потиска
споделяне на функцията на някои лимфоцитни клонове, по-специално syn
тези, които синтезират леките вериги на имуноглобулините, участващи в
образуване на амилоидни фибрили (това също се отнася до
амилоидоза, развиваща се при миелома).
Въздействието върху основните клинични синдроми включва
премахване на отоци, хипертония, както и мерки, насочени към
борба с развитието на бъбречна недостатъчност:
а) с развитието на нефротичен синдром и тежък оток
необходимо е достатъчно протеиново съдържание в храната, намаляване на
варена сол, както и въвеждането на цяла кръв или червени кръвни клетки
маса (особено при наличие на анемия), внимателна употреба
прием на диуретици;
б) артериалната хипертония е необичайна при амилоидоза,
обаче, когато достигне високи числа, е необходимо да се предпише
въвеждане на различни видове антихипертензивни лекарства;
в) ако се развие бъбречна недостатъчност, лечението се провежда в съответствие с общоприетия план (ограничаване на протеина в храната, прием на достатъчно течности, корекция на минералния метаболизъм). При бъбречна недостатъчност, причинена от амилоидоза, са възможни хемодиализа и бъбречна трансплантация.
Прогноза.Продължителността на протеинуричния период е трудно да се определи, но след откриването му отокът обикновено се развива след 3 години, на фона на който бързо възниква хронична бъбречна недостатъчност. Всичко това прави прогнозата доста сериозна.
Предотвратяване.За идиопатичната и генетичната амилоидоза мерките за първична превенция не са известни. В случай на вторична амилоидоза профилактиката се състои в лечение на заболявания, водещи до развитие на амилоидоза.
RCHR (Републикански център за развитие на здравеопазването към Министерството на здравеопазването на Република Казахстан)
Версия: Клинични протоколи на Министерството на здравеопазването на Република Казахстан - 2016 г
Амилоидоза (E85)
Нефрология
Главна информация
Кратко описание
Одобрено
Съвместна комисия по качество на здравеопазването
Министерство на здравеопазването и социалното развитие на Република Казахстан
от 13 октомври 2016 г
Протокол No13
Амилоидоза- група заболявания, чийто отличителен белег е отлагането на фибриларен гликопротеин - амилоид - в тъканите и органите.
Корелация на кодовете по МКБ-10 и МКБ-9
| МКБ-10 | МКБ-9 | ||
| Код | Име | Код | Име |
| E85 | Амилоидоза |
55.23 99.76 |
Затворена [перкутанна] [пункционна] бъбречна биопсия. Хемодиализа. Терапевтична плазмафереза Екстракорпорална имуноадсорбция |
| E85.0 | Наследствена фамилна амилоидоза без невропатия | ||
| E85.1 | Невропатична наследствена амилоидоза | ||
| E85.2 | Наследствена амилоидоза, неуточнена | ||
| E85.3 | Вторична системна амилоидоза | ||
| E85.4 | Ограничена амилоидоза | ||
| E85.8 | Други форми на амилоидоза | ||
| E85.9 | Амилоидоза, неуточнена | ||
Дата на разработване/ревизиране на протокола: 2016 г.
Потребители на протокола:Общопрактикуващи лекари, терапевти, хематолози, нефролози.
Скала на нивото на доказателства:
| А | Висококачествен мета-анализ, систематичен преглед на RCT или големи RCT с много ниска вероятност (++) за отклонение, резултатите от които могат да бъдат обобщени за подходяща популация. |
| IN | Висококачествен (++) систематичен преглед на кохортни или случай-контролни проучвания, или висококачествени (++) кохортни или случай-контролни проучвания с много нисък риск от отклонение, или РКИ с нисък (+) риск от отклонение, резултатите от които могат да бъдат обобщени за подходяща популация. |
| СЪС |
Кохортно проучване или проучване случай-контрола или контролирано изпитване без рандомизация с нисък риск от отклонение (+). Резултатите от които могат да бъдат обобщени за съответната популация или RCTs с много нисък или нисък риск от отклонение (++ или +), резултатите от които не могат да бъдат директно обобщени за съответната популация. |
| д | Серия от случаи или неконтролирано проучване или експертно мнение. |
Класификация
Видове амилоид и съответните форми на амилоидоза:
|
Протеин амилоид |
Прекурсорен протеин | Клинична форма на амилоидоза |
| АА | SAA протеин | Вторична амилоидоза при хронични възпалителни заболявания, включително периодично заболяване и синдром на Muckle-Wells |
| АЛ | λ, κ-леки вериги на имуноглобулини | Амилоидоза при плазмоклетъчни дискразии - идиопатична, при мултиплен миелом и макроглобулинемия на Waldenström |
| ATTR | Транстиретин | Фамилни форми на полиневропатична, кардиопатична и друга амилоидоза, системна сенилна амилоидоза |
| Ар2М | β2-микроглобулин | Диализна амилоидоза |
| AGel | Гелсолин | Финландска фамилна амилоидна полиневропатия |
| AApoAI | Аполипопротеин A-I | Амилоидна полиневропатия (тип III, според van Allen, 1956) |
| AFib | Фибриноген | Амилоидна нефропатия |
| Ар | β протеин | Болест на Алцхаймер, синдром на Даун, наследствени мозъчни кръвоизливи с амилоидоза, Холандия |
| APrP Scr | Прионов протеин | болест на Кройцфелд-Якоб, болест на Герстман-Щрауслер-Шайнкер |
| AANF | Предсърден натриуретичен фактор | Изолирана предсърдна амилоидоза |
| AIAPP | Амилин | Изолирана амилоидоза в Лангерхансовите острови с захарен диабетТип II, инсулином |
| ACal | Прокалцитонин | За медуларен рак щитовидната жлеза |
| ACys | Цистатин С | Наследствени мозъчни кръвоизливи с амилоидоза, Исландия |
Клинична класификация на амилоидоза
първична амилоидоза:
· възникващи без видима причина;
свързани с мултиплен миелом;
вторична амилоидоза:
· при хронични инфекции;
· при ревматоиден артрит и други заболявания на съединителната тъкан;
· при онкологични заболявания;
фамилна (наследствена) амилоидоза:
· с периодично боледуване;
· португалски вариант и други форми на фамилна амилоидоза;
сенилна амилоидоза
локална амилоидоза
наследствена амилоидоза:
невропатичен
· с увреждане на долните крайници: португалски, японски, шведски и други видове;
· с увреждане на горните крайници: типове Швейцария-Индиана, Германия-Мериленд;
нефропатичен:
· периодично боледуване;
· температура и болки в корема при шведи и сицилианци;
комбинация от обрив, глухота и увреждане на бъбреците;
· бъбречно увреждане в комбинация с артериална хипертония;
кардиомиопатичен:
Датски - прогресираща сърдечна недостатъчност;
· Мексиканско-американски - синдром на болния синус, предсърден арест;
смесени:
Финландски - дистрофия на роговицата и увреждане на черепномозъчните нерви;
· мозъчни инсулти.
Клинични стадии на бъбречна амилоидоза
| сцена | Клинична изява |
| 1 | Предклиничен или латентен (асимптоматичен) стадий - амилоидът присъства в междинната зона и се развиват отоци и огнища на склероза по протежение на правите съдове на пирамидите. Етапът продължава 3-5 или повече години. През този период при реактивна амилоидоза преобладават клиничните прояви на основното заболяване (например, гноен процес в белите дробове, туберкулоза, ревматоиден артрит и др.). |
| 2 | Протеинурен (албуминуричен) стадий - амилоидът се появява предимно в мезангиума, в капилярните бримки, в пирамидите и кората на гломерулите, в съдовете. Развиват се склероза и атрофия на нефрона, хиперемия и лимфостаза. Пъпките са уголемени и плътни, матово сиво-розови на цвят. Протеинурията в началото е умерено изразена, може дори да е преходна за известно време, да намалява и да се увеличава, но след това става персистираща (стадий на интермитентна протеинурия). Някои изследователи разграничават два периода на този етап: селективна и неселективна протеинурия. Продължителността на етапа е от 10 до 13 години. |
| 3 | Нефротичен (едематозен, едематозно-хипотоничен) стадий - амилоидно-липоидна нефроза - амилоид във всички части на нефрона. Има склероза и амилоидоза на медулата, но кортикалния слой е без изразени склеротични промени. Продължителността на етапа е до 6 години. И в протеинуричния, и в нефротичния стадий бъбреците са уголемени и плътни (голям мастен бъбрек). Клинично този етап се проявява с класически нефротичен синдром с всичките му признаци: с развитие на масивна протеинурия (със загуба на протеин в урината повече от 3-5 грама на ден), хипопротеинемия с хипоалбуминемия, хиперхолестеролемия, липидурия с оток до степента на анасарка. В уринарния седимент се откриват хиалинни отливки, а с увеличаване на протеинурията се откриват гранулирани отливки. Възможни са микро- и макрохематурия и левкоцитурия без признаци на пиелонефрит. |
| 4 | Уремичен (терминален, азотемичен) стадий - амилоидно набръчкан бъбрек - намален по размер, плътен, с белези. Хроничната бъбречна недостатъчност се различава малко от тази при други бъбречни заболявания. Смята се, че за разлика от гломерулонефрита, при който появата на хронична бъбречна недостатъчност, протичаща с полиурия, може да доведе до поне частична конвергенция на отока, при амилоидоза азотемията се развива на фона на ниско кръвно налягане и нефротичен синдром. |
Диагностика (амбулатория)
АМБУЛАТОРНА ДИАГНОСТИКА
Диагностични критерии
Оплаквания:
слабост, повишена умора;
· главоболие;
· подуване на краката, ръцете и лицето;
· високо кръвно налягане;
Гадене, диария (диария);
· болки в сърдечната област;
· болка в мускулите.
Анамнеза:
· отслабване;
· наличие на моноклонална гамапатия с неясен произход;
· хронични възпалителни (гнойни) заболявания;
· хронични инфекции;
· наследственост.
Физическо изследване
Обща проверка:
· периорбитална пурпура (наблюдава се в 15% от случаите);
Макроглосията е характерна за първичната амилоидоза (AL);
· задух при физическо усилие (наблюдава се при около 40% от пациентите);
· раменен знак (периартикуларната инфилтрация на амилоид води до фалшива хипертрофия и увеличаване на обема на мускулите на раменния пояс и бедрото).
Аускултация:
Може да има нарушение на сърдечния ритъм.
палпация:
· оток на долните крайници, дължащ се на хипоалбуминемия и нефротичен синдром, както и поради стагнация в системното кръвообращение поради рестриктивна кардиомиопатия (наблюдавана в 50% от случаите);
Увеличен черен дроб и далак;
· парестезия (наблюдава се при около 15% от пациентите);
· спазматична болка в стомашно-чревния тракт;
· може да има увеличение на субмандибуларните слюнчени жлези.
Лабораторни изследвания:
· пълна кръвна картина - анемия, левкоцитоза, повишена СУЕ;
· общ анализ на урината - протеинурия, микрохематурия, асептична левкоцитурия;
· биохимичен кръвен тест (общ протеин, албумин, Na, Ca, холестерол, захар в кръвния серум) - хипопротеинемия (поради хипоалбуминемия), хиперглобулинемия, хипонатриемия, хипопротромбинемия, хипокалцемия, хиперхолестеролемия.
· Ехография на органи коремна кухинаи бъбреци - визуализират се увеличени, уплътнени бъбреци (големи мастни бъбреци).

Диагностика (болница)
ДИАГНОСТИКА НА СТАЦИОНАРНО НИВО
Диагностични критерии на болнично ниво
Оплаквания и анамнеза:вижте амбулаторно ниво.
Физическо изследване:вижте амбулаторно ниво.
Лабораторни изследвания:
| Диагностичен тест | Резултат |
|
Серумна имунофиксация Тестът е положителен при 60% от пациентите с имуноглобулинова лека верига (AL) амилоидоза (6). |
|
|
Имунофиксация на урината Тестът е положителен при 80% от пациентите с AL амилоидоза (6). Откриването на белтък от лека верига в урината предполага наличието на мултиплен миелом и амилоидоза. |
Наличие на моноклонален протеин |
|
Тест за свободни имуноглобулини с лека верига в серума Този сравнително нов тест с много висока чувствителност(>95%) за диагнозата AL амилоидоза (10). Обикновено се използват налични в търговската мрежа антисеруми срещу леки вериги на имуноглобулини, AA и транстиретин, но може да им липсва достатъчна специфичност и чувствителност. В много случаи масспектроскопията и имуноелектронната микроскопия са необходими за определяне на подлежащия тип амилоид. |
Ненормално капа ламбда съотношение |
|
Биопсия костен мозък
Биопсия на костен мозък се извършва при всички пациенти със съмнение за амилоидоза на леката верига и се страхотен източниктъкан за диагностициране на всеки пациент със съмнение за амилоидоза. |
Наличие на клонални плазмени клетки |
Инструментални изследвания:
· Ехография на коремни органи и бъбреци - визуализират се уголемени, уплътнени бъбреци (големи мастни бъбреци).
Алгоритъм за диагностика на бъбречна амилоидоза
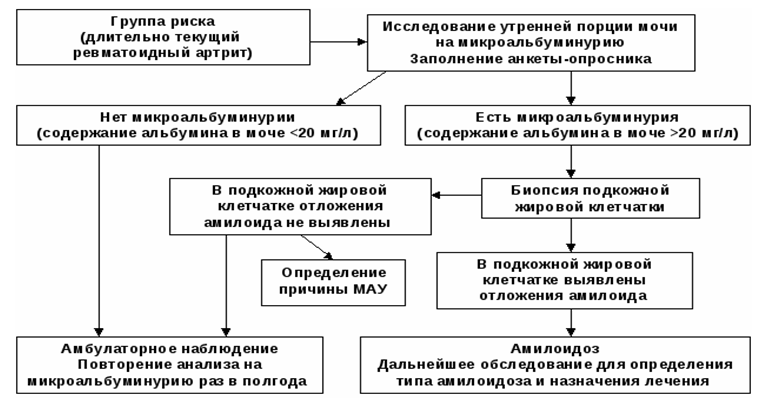
Списък на основните диагностични мерки:
· общ кръвен анализ;
· общ анализ на урината;
· биохимичен кръвен тест (общ белтък, албумин, Na, Ca, холестерол, кръвна захар);
Серумна имунофиксация;
· имунофиксация на урината;
· изследване на свободни имуноглобулини с лека верига в серума;
Биопсия на костен мозък.
· Ехография на коремни органи и бъбреци.
Списък на допълнителни диагностични мерки
Лабораторни изследвания:
| Диагностичен тест | Резултат |
|
Тъканна биопсия: За диагностициране на амилоидоза тъканните отлагания в биопсичния материал трябва да бъдат оцветени положително с конго червено (11). Ярко зелено двойно пречупване може да се види, когато материалът Конго е оцветен в червено под поляризирана светлина. Материал за биопсия може да се вземе от лигавицата на устните, кожата, венците, подкожната мазнина, костния мозък, нервите, ректума, бъбреците, черния дроб или сърцето. Отлаганията винаги са разположени извънклетъчно и са аморфни. |
положително - зелено двойно пречупване при оцветяване с конго червено |
|
Имунохистологични изследвания на амилоидни отлагания: Те позволяват разпознаването на различни форми на системна амилоидоза. |
антисерум към имуноглобулинова лека верига, AA и транстиретин |
|
Масспектроскопия: Предоставя анализ на състава на амилоидния протеин. В момента златният стандарт за диагностициране на типа амилоидоза. |
потвърждава вида на протеина |
|
Имуноелектронна микроскопия: Всички форми на амилоид електронен микроскопвлакнеста, жилава и неразклонена. |
амилоидите имат фибриларен вид и са твърди и неразклонени. |
|
Генетично изследване: За да се изключи наследствена амилоидоза в случай на съмнителни резултати от теста за откриване на моноклонален имуноглобулинов протеин/свободна амилоидна лека верига, генетичното изследване е задължително. Гените могат да бъдат изследвани чрез директно секвениране и включват следните гени: TTR, фибриноген, аполипопротеин А1, лизозим, MEFV (средиземноморска треска) и рецептор на тумор некрозис фактор 1 (TNFR1 или TNFRSF1A). MEFV и TNFRSF1A са наследствени синдроми периодична треска(т.е. потенциални причини за АА амилоидоза) и не са наследствени амилоидози сами по себе си. |
положителен |
|
Сцинтиграфско сканиране на серумен амилоид P (SAP): През последните години сцинтиграфия с белязан с йод серумен P компонент (SAP) се използва в клиничната практика за оценка на разпределението на амилоид в тялото. |
поглъщане в местата на отлагане на амилоид |
|
Общ анализкръв: Анемията се наблюдава главно при пациенти с бъбречна недостатъчност или кървене от стомашно-чревния тракт. Тромбоцитемията е свързана със засягане на черния дроб и хиперспленизъм. |
обикновено нормално |
|
Биохимичен кръвен тест (черен дроб и бъбреци), показатели за метаболитен статус): Амилоидозата на черния дроб се характеризира с повишаване на нивото алкална фосфатаза. Повечето пациенти с ранен стадий на бъбречна амилоидоза поддържат креатининов клирънс, но могат да получат значителна степен на хипоалбуминемия поради загуба на протеин в урината (нефротичен синдром). |
Ниски нива на албумин; повишаване на алкалната фосфатаза |
|
Ежедневна протеинурия (събиране на урина за 24 часа): Екскреция на албумин > 1 g/ден при пациенти с амилоидоза показва увреждане на бъбреците (бъбречна амилоидоза). При нива на протеинурия > 3 g/ден се развива нефротичен синдром. |
повишен протеин в урината |
|
Ниво на серумен тропонин: Чувствителен тест за определяне на увреждане на миокарда. Пациентите с откриваеми нива на тропонин имат по-лоша прогноза от тези без такива (12). |
повишена |
|
B-тип натриуретичен пептид: Чувствителен диагностичен тест за наличие на разтягане на миокарда и ХСН. Доказано е, че е важно прогностична стойностпри установяване на сърдечна амилоидоза (13). При нива на натриуретичен пептид тип B >300 ng/L (>300 pg/ml) предполага засягане на миокарден амилоид (10). Пациенти със<170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с >170 ng/l (>170 pg/ml). |
повишена |
|
бета-2 микроглобулини: Той е предиктор за преживяемостта при пациенти с амилоидоза. При ниво на бета-2-микроглобулин > 2,7 mg/l прогнозата е неблагоприятна (14). |
повишена |
Инструментални изследвания:
|
ЕКГ: Трябва да се извършва при всички пациенти като част от оценката на сърдечното засягане. |
нарушение на проводимостта на сърцето |
|
Ехокардиограма (EchoCG): Клиничните признаци на сърдечна недостатъчност при пациенти със сърдечна амилоидоза се срещат при 22% до 34% (7). Ехокардиографията разкрива висока честота на отлагане на амилоид при пациенти с минимални симптоми (около 50% от случаите на АЛ имат сърдечно засягане). IN последен етапима намаляване на фракцията на изтласкване. |
диастолна дисфункция, удебеляване на междукамерната преграда, намалена фракция на изтласкване |
|
Доплерово ехо с напрежение: Индикатор за степента на амилоидна инфилтрация в миокарда. Има висока чувствителност за откриване на аномалии, когато няма хипертония или клапно сърдечно заболяване. Разтягането на миокарда се дефинира като процентна промяна в дължината на миокардните влакна за единица дължина, а скоростта зависи от продължителността на разтягането (15–16). |
Намаляване на надлъжната контракция и разтягане на миокарда; ограничаване на вентрикуларното пълнене |
|
ЯМР на сърцето: Магнитно-резонансната релаксометрия подобрява надеждността на MRI диагностиката и помага да се разграничи сърдечната амилоидоза от хипертрофичната кардиомиопатия. |
значително увеличени времена за релаксация Т1 и Т2 в сравнение с контролите, свързани с възрастта |
Диференциална диагноза
Диференциална диагнозабъбречна амилоидоза
| състояние | Различни признаци/симптоми | Диференцируеми тестове |
| Хипертрофична кардиомиопатия | Клинично е трудно да се разграничи HCM от сърдечна амилоидоза. |
· Ехокардиография е диагностичен критерийза GCM, където се открива асиметрична хипертрофия на интервентрикуларната преграда; · волтажното доплерово ехо, използвано за изключване на признаци на амилоидоза, не показва типичните рестриктивни промени в пълненето, открити при амилоидоза; · MRI позволява да се разграничат 2 синдрома |
| Мембранозна гломерулопатия | Клинично идентични прояви при пациенти с нефротичен синдром. | · бъбречна биопсия не се оцветява с конго червено. |
| Невропатия, свързана с моноклонална гамапатия с неизвестен произход (MGUS). | Пациентите нямат значима протеинурия, хепатомегалия или кардиомиопатия. | · биопсията на суралния нерв не се оцветява с конго червено. |
| Множествена миелома | Болки в костите, симптоми на анемия и бъбречна недостатъчност. |
· обикновени рентгенови лъчипоказват литични костни лезии, компресионни фрактури, дифузна остеопороза; · нисък Hb; · бъбречна недостатъчност. |
| Нефротичен синдром | Дневна протеинурия над 3,5 g/ден, оток, хипоалбуминемия, дислипидемия | · виж КП „Нефротичен синдром“ |
Медицински туризъм
Лекувайте се в Корея, Израел, Германия, САЩ
Медицински туризъм
Получете съвет за медицински туризъм
Лечение
внимание!
- Като се самолекувате, можете да причините непоправима вреда на здравето си.
- Информацията, публикувана на уебсайта на MedElement, не може и не трябва да замества консултация лице в лицелекар Не забравяйте да се свържете лечебни заведенияако имате някакви заболявания или симптоми, които Ви притесняват.
- Изборът на лекарства и тяхната дозировка трябва да се обсъди със специалист. Само лекар може да предпише правилното лекарство и неговата дозировка, като вземе предвид заболяването и състоянието на тялото на пациента.
- Уебсайтът на MedElement е единствено информационен и справочен ресурс. Информацията, публикувана на този сайт, не трябва да се използва за неоторизирана промяна на лекарски предписания.
- Редакторите на MedElement не носят отговорност за каквито и да е лични наранявания или имуществени щети в резултат на използването на този сайт.
- общ, системно заболяванеорганизъм, при който отлагането на специфичен гликопротеин (амилоид) се извършва в органи и тъкани с нарушена функция на последния. При амилоидоза могат да бъдат засегнати бъбреците (нефротичен синдром, едематозен синдром), сърцето (сърдечна недостатъчност, аритмии), стомашно-чревния тракт, опорно-двигателния апарат и кожата. Възможно развитие на полисерозит, хеморагичен синдром, психични разстройства. Надеждната диагноза на амилоидозата се улеснява от откриването на амилоид в проби от биопсия на засегнатите тъкани. За лечение на амилоидоза се провежда имуносупресивна и симптоматична терапия; по показания - перитонеална диализа, бъбречна и чернодробна трансплантация.
МКБ-10
E85

Главна информация
Амилоидозата е заболяване от групата на системните диспротеинози, протичащо с образуването и натрупването в тъканите на сложно протеиново-полизахаридно съединение - амилоид. Разпространението на амилоидозата в света до голяма степен се определя географски: например периодичното заболяване е по-често срещано в страните от средиземноморския басейн; амилоидна полиневропатия – в Япония, Италия, Швеция, Португалия и др. Средна честотаамилоидоза в населението е 1 случай на 50 хил. население. Заболяването обикновено се развива при хора на възраст над 50-60 години. Като се има предвид факта, че амилоидозата засяга почти всички органи и системи, заболяването се изучава от различни медицински дисциплини: ревматология, урология, кардиология, гастроентерология, неврология и др.
Причини за амилоидоза
Етиологията на първичната амилоидоза не е напълно проучена. Въпреки това е известно, че вторичната амилоидоза обикновено се свързва с хронични инфекциозни (туберкулоза, сифилис, актиномикоза) и гнойно-възпалителни заболявания (остеомиелит, бронхиектазии, бактериален ендокардит и др.), По-рядко - туморни процеси(лимфогрануломатоза, левкемия, рак на висцералните органи). Реактивна амилоидоза може да се развие при пациенти с атеросклероза, псориазис, ревматоиден артрит (ревматоиден артрит, анкилозиращ спондилит), хронично възпаление(неспецифичен улцерозен колит, болест на Crohn), мултисистемни лезии (болест на Whipple, саркоидоза). Сред факторите, допринасящи за развитието на амилоидоза, от първостепенно значение са хиперглобулинемията, нарушеното функциониране на клетъчния имунитет, генетичната предразположеност и др.
Патогенеза
Сред многото версии на амилоидогенезата най-голямото числоТеориите за диспротеинозата, локалния клетъчен генезис, имунологичните и мутационните теории имат поддръжници. Теорията за локалния клетъчен генезис разглежда само процеси, протичащи на клетъчно ниво (образуването на фибриларни амилоидни прекурсори от макрофагалната система), докато образуването и натрупването на амилоид се извършва извън клетката. Следователно теорията за локалния клетъчен генезис не може да се счита за изчерпателна.
Според теорията за диспротеинозата амилоидът е продукт на анормален протеинов метаболизъм. Основните връзки в патогенезата на амилоидозата - диспротеинемия и хиперфибриногенемия - допринасят за натрупването на груби фракции на протеини и парапротеини в плазмата. Имунологичната теория за произхода на амилоидозата свързва образуването на амилоид с реакцията антиген-антитяло, при която антигените са чужди протеини или продукти на разпадане на собствените тъкани. В този случай отлагането на амилоид се проявява предимно в областите на образуване на антитела и излишни антигени. Най-универсалната е мутационната теория на амилоидозата, която взема предвид огромно разнообразие от мутагенни фактори, които могат да причинят анормален протеинов синтез.
Амилоидът е сложен гликопротеин, състоящ се от фибриларни и глобуларни протеини, тясно свързани с полизахариди. Амилоидните отлагания се натрупват в интимата и адвентицията на кръвоносните съдове, стромата на паренхимните органи, жлезистите структури и др. При незначителни амилоидни отлагания промените се откриват само на микроскопично ниво и не водят до функционални нарушения. Тежкото натрупване на амилоид е придружено от макроскопски промени в засегнатия орган (увеличен обем, мазен или восъчен вид). В резултат на амилоидоза се развива стромална склероза и атрофия на паренхима на органа и тяхната клинично значима функционална недостатъчност.
Класификация
Според причините се разграничават първична (идиопатична), вторична (реактивна, придобита), наследствена (фамилна, генетична) и сенилна амилоидоза. Има различни форми на наследствена амилоидоза: средиземноморска треска или периодично заболяване (пристъпи на треска, коремна болка, запек, диария, плеврит, артрит, кожни обриви), португалска невропатична амилоидоза (периферна полиневропатия, импотентност, нарушения на сърдечната проводимост), финландски тип ( атрофия на роговицата, черепна невропатия), датски вариант (кардиопатична амилоидоза) и много други. и т.н.
В зависимост от преобладаващото увреждане на органите и системите, нефропатична (амилоидоза на бъбреците), кардиопатична (амилоидоза на сърцето), невропатична (амилоидоза на нервната система), хепатопатична (амилоидоза на черния дроб), епинефропатична (амилоидоза на надбъбречните жлези). ), APUD-амилоидоза, амилоидоза на кожата и смесен тип заболяване. Освен това в международната практика е обичайно да се прави разлика между локална и генерализирана (системна) амилоидоза. Към локализирани форми, обикновено развиващи се при индивиди старост, включват амилоидоза при болестта на Алцхаймер, захарен диабет тип 2, ендокринни тумори, кожни тумори, Пикочен мехури т.н. В зависимост от биохимичен съставСред системните форми на амилоидоза се разграничават следните видове амилоидни фибрили:
- АЛ- като част от фибрили, Ig леки вериги (с болест на Waldenström, множествена миелома, злокачествени лимфоми);
- А.А.- като част от фибрилите, острофазов серумен α-глобулин, подобен по своите характеристики на С-реактивния протеин (при туморни и ревматични заболявания, периодични заболявания и др.);
- Ар2М- като част от фибрилите β2-микроглобулин (при хронична бъбречна недостатъчност при пациенти на хемодиализа);
- ATTR- в състава на фибрилите, транспортния протеин транстиретин (при фамилни наследствени и сенилни форми на амилоидоза).
Симптоми на амилоидоза
Клиничните прояви на амилоидозата са разнообразни и зависят от тежестта и локализацията на амилоидните отлагания, биохимичния състав на амилоида, "стажа" на заболяването и степента на органна дисфункция. В латентния стадий на амилоидозата, когато амилоидните отлагания могат да бъдат открити само микроскопски, няма симптоми. Тъй като функционалната недостатъчност на даден орган се развива и прогресира, клиничните признаци на заболяването се увеличават.
При бъбречна амилоидоза дългосрочният стадий на умерена протеинурия се заменя с развитието на нефротичен синдром. Преходът към напреднал стадий може да бъде свързан с интеркурентна инфекция, ваксинация, хипотермия или обостряне на основното заболяване. Отокът постепенно се увеличава (първо в краката, а след това в цялото тяло), развива се нефрогенна артериална хипертония и бъбречна недостатъчност. Може да възникне тромбоза на бъбречната вена. Масивната загуба на протеин е придружена от хипопротеинемия, хиперфибриногенемия, хиперлипидемия и азотемия. В урината се откриват микро-, понякога макрохематурия и левкоцитурия. Като цяло, по време на бъбречна амилоидоза се разграничават ранен неедематозен стадий, едематозен стадий и уремичен (кахектичен) стадий.
Сърдечната амилоидоза протича като рестриктивна кардиомиопатия с характерни клинични прояви - кардиомегалия, аритмия, прогресираща сърдечна недостатъчност. Пациентите се оплакват от задух, подуване и слабост, които се появяват при незначително физическо натоварване. По-рядко при сърдечна амилоидоза се развива полисерозит (асцит, ексудативен плеврит и перикардит).
Увреждането на стомашно-чревния тракт при амилоидоза се характеризира с амилоидна инфилтрация на езика (макрогласия), хранопровода (ригидност и нарушена перисталтика), стомаха (киселини, гадене), червата (запек, диария, синдром на малабсорбция, чревна обструкция). Стомашно-чревно кървене може да се появи на различни нива. При амилоидна инфилтрация на черния дроб се развива хепатомегалия, холестаза и портална хипертония. Увреждането на панкреаса поради амилоидоза обикновено се прикрива като хроничен панкреатит.
Кожната амилоидоза се проявява с появата на множество восъчни плаки (папули, възли) по лицето, шията и естествените кожни гънки. По външни признаци кожните лезии могат да наподобяват склеродермия, невродермит или лихен планус. За амилоидни лезии на опорно-двигателния апарат, развитието на симетричен полиартрит, карпален тунелен синдром, гленохумерален периартрит, миопатия. Индивидуални формиамилоидозата, засягаща нервната система, може да бъде придружена от полиневропатия, парализа на долните крайници, главоболие, световъртеж, ортостатична хипотония, изпотяване, деменция и др.
Диагностика
С клиничните прояви на амилоидозата могат да се сблъскат различни клиницисти: ревматолози, уролози, кардиолози, гастроентеролози, невролози, дерматолози, терапевти и др. От първостепенно значение за правилна настройкаДиагнозата се основава на цялостна оценка на клиничните и анамнестичните признаци и цялостно лабораторно и инструментално изследване.
Амилоидозата е системно заболяване, засяга протеинов метаболизъм, имунната система спира да работи. В тази връзка се образува амилоид - протеиново-захариден комплекс, който се отлага във всички тъкани на човешките органи.
С течение на времето амилоидът все повече засяга органите, измествайки нормалните клетки. В резултат на това органът губи функционалност и необратими промени. Ако болестта не се лекува за дълго време, функциите на няколко органа са нарушени, което води до смърт.
Според изследванията на СЗО амилоидозата се диагностицира при приблизително 1% от жителите на света. Вторичната амилоидоза е най-честата. Генетичната амилоидоза се диагностицира по-често при хора от еврейска, арменска националност, както и при жители на средиземноморските страни.
Заболеваемостта при мъжете е два пъти по-висока от тази при жените. Сред всички форми на амилоидоза се диагностицира нефропатична (увреждане на бъбреците) и генерализирана (увреждане на всички тъкани и органи) амилоидоза.
Видове амилоидоза, причини за развитие
В зависимост от причината за амилоидозата има видове заболявания, които могат да се развият самостоятелно или поради патологии в други системи и органи. Среща следните видовеамилоидоза: сенилна, с тумори, първична или идиопатична амилоидоза, наследствена, вторична или реактивна, както и при пациенти на хемодиализа. В зависимост от вида, развитието на амилоидоза протича по различен начин, симптомите и прогнозата варират. По-долу ще разгледаме подробно видовете и етапите на амилоидозата.
Първичен (идиопатичен)
Първичната амилоидоза в повечето случаи започва без причина. При тази форма на заболяването амилоидът се отлага в тъканите и органите и се наблюдава мутация на клетките на имунната система. AL-амилоидът, образуван в процеса, се натрупва в мускулите, кожата, сърдечно-съдовата система и нервите. Също така, AL амилоидът се образува на фона на туморен миелом, когато плазмените клетки започват да секретират глобулини в големи количества. След свързване с плазмените нуклеопротеини анормалните глобулини се превръщат в амилоид.
Вторичен (реактивен)
Вторичната амилоидоза се развива на фона на прогресиращи възпалителни процеси във времето. В този случай АА амилоидът се образува като усложнение на други заболявания. Причините за възникване на вторична амилоидоза са:
- Хронични инфекции - проказа, малария, туберкулоза, сифилис, пиелонефрит, бронхиектазии.
- Гнойни хронични заболявания - нагнояване на рани за дълго време, остеомиелит.
- Тумори – левкемия, лимфогрануломатоза и др.
- Наличието на неспецифичен улцерозен колит (възпаление на дебелото черво).
- Ревматологични заболявания – анкилозиращ спондилит, ревматоиден артрит и др.
Вторичната амилоидоза може да засегне всеки орган или тъкан в тялото. Проявата на заболяването не се забелязва веднага. Години след началото на основното заболяване може да се забележи дисфункция на органа, където се отлага най-много амилоид. Най-често от това заболяване се засягат черният дроб, бъбреците, далака и лимфните възли. С течение на времето други органи са засегнати, което води до полиорганна недостатъчност и смърт.
Наследствена амилоидоза
Наследствената форма на амилоидоза се причинява от наличието на мутирали гени в клетките на имунната система. Тези генетични мутации се предават през поколенията, което води до образуването на амилоидобласти. Наследствената форма засяга хора в определена област или принадлежащи към определена етническа група. Наследствената амилоидоза се разделя на видове:
- Кардиопатичен. Основно се диагностицира при жители на Дания. Клиничната картина на заболяването прилича на първична амилоидоза от генерализиран тип.
- Невропатична. Характеризира се с поражение нервна тъкан. В зависимост от местоположението на лезията се различават португалска (нерви на краката), американска (нерви на ръцете), финландска (нервна система, роговица, бъбреци) амилоидоза.
- Фамилна нефропатия. Друго име е английската амилоидоза (болест на Мукъл и Уелс). Клиничната картина е уртикария, пристъпи на треска, увреждане на слуха.
- Периодично (фамилна средиземноморска треска). Заболяването е по-разпространено сред евреи, араби и арменци. Прояви – температура над 39ºС, болка в главата и мускулите, обилно изпотяване. Наблюдава се възпаление на мембраните на белите дробове, перитонеалните органи и синовиалните органи. Психичните аномалии са чести.
Сенилна амилоидоза
При хора, навършили 80 години, амилоидът се отлага локално в различни тъкани и органи. Заболяването е свързано с други заболявания, свързани с възрастта. Има два вида сенилна амилоидоза:
- Церебрална или церебрална. Развива се на фона на болестта на Алцхаймер. Амилоидът Ab се отлага в мозъчната тъкан.
- Сърдечно. Може да засегне вентрикулите на сърцето (когато амилоидът се образува от мутирал кръвен протеин транстиретин) и предсърдията (когато амилоидът се образува от натриуретичен пептид, секретиран от сърдечните клетки). И в двата случая амилоидите се откриват в тъканите на белите дробове, панкреаса и далака.
За тумори
Някои видове тумори засягат злокачествената трансформация на клетките на болния орган, които в резултат на това произвеждат фибриларен протеин. В този случай амилоидозата се развива локално в тъканта на органа, засегнат от тумора. Причини, които провокират амилоидоза при тумори:
- Медуларен тумор на щитовидната жлеза. Ракът се развива от С клетките на щитовидната жлеза, които в добро състояниеотговорен за производството на калцитоцин. Когато синтезата на калцитоцин е нарушена, неговите фрагменти стават част от амилоида AE.
- Рак на островчетата на щитовидната жлеза. Островчетата са клъстери от клетки, отговорни за производството на хормони - глюкагон, инсулин, соматостатин и др. Злокачествената дегенерация на клетките предизвиква освобождаване на фибриларен протеин, който впоследствие се дегенерира в амилоид.
Амилоидоза при хемодиализа
 Хемодиализата е животоспасяващо лечение за пациенти, чиито бъбреци не са в състояние да премахнат токсините от кръвта. странични продуктиметаболизъм. Хемодиализата се предписва при диагностицирани с бъбречна недостатъчност (остра, хронична).
Хемодиализата е животоспасяващо лечение за пациенти, чиито бъбреци не са в състояние да премахнат токсините от кръвта. странични продуктиметаболизъм. Хемодиализата се предписва при диагностицирани с бъбречна недостатъчност (остра, хронична).
Същността на процедурата е преминаването на кръвта през машина, която премахва вредните вещества от нея, връщайки пречистената кръв в тялото на пациента.
По време на диализа В2-микроглобулинът не може да бъде отстранен от тялото и ако пациентът дълго времепринудени да преминат хемодиализа, протеинът се натрупва в тялото в прекомерни количества. Той се свързва с плазмените нуклеопротеини и се установява различни органи, става основата на амилоида.
Симптоми на амилоидоза
Като се има предвид, че болестта може да се разпространи във всеки орган или тъкан, симптомите ще варират. Различни формиБолестите в началото на своето протичане се характеризират с увреждане и дисфункция на един орган в човешкото тяло.
С течение на времето заболяването (ако не е локална амилоидоза) прогресира, засягайки други органи и тъкани. Прояви на амилоидоза могат да се наблюдават в бъбреците, черния дроб, сърцето и надбъбречните жлези, далака, стомашно-чревния тракт и нервната система, ставите, мускулите и кожата. Видовете заболяване са описани подробно по-долу.
Бъбречно увреждане
Най-често се счита за бъбречна амилоидоза опасна болест, в сравнение с увреждане на други органи. Клиничната картина на бъбречната амилоидоза зависи от етапа. Общо има 4 от тях - латентна, нефротична, азотемична, протеинурична.
В латентния стадий бъбречната амилоидоза практически не показва симптоми. Ако това е вторична форма, пациентът усеща симптомите на основното заболяване. Само години по-късно бъбречното увреждане ще стане симптоматично.
В протеинуричния стадий бъбречната амилоидоза продължава 10 или повече години. По това време амилоидозата постепенно се отлага в съдовете, междуклетъчното пространство и гломерулите на бъбреците. Поради това нефроните, които произвеждат урина, се компресират, атрофират и умират. Целостта на бъбречния филтър, който обикновено не позволява преминаването на големи молекулни протеини и кръвни клетки, е нарушена. Впоследствие протеините се екскретират в урината. На този етап е трудно да се подозира амилоидоза на бъбреците, тъй като екскреторната функция не е нарушена. Проблемът може да бъде открит в резултатите от лабораторните изследвания.
Бъбречната амилоидоза в нефротичния стадий се проявява чрез по-нататъшно разрушаване на бъбречния филтър. Поради това голямо количество протеин се губи с урината, концентрацията му в кръвта намалява. Протеините са компонент на процеса на задържане на кръвта в кръвоносните съдове. С намаляване на концентрацията на протеини течността навлиза в тъканите, подуване се появява по всяко време на деня, независимо от позицията на тялото. Освен това бъбречната амилоидоза прогресира, отокът е тежък. Течността се натрупва в перитонеума, плевралната кухина и сърдечната торбичка. Този етап продължава 4-6 години.
На азотемичен етап от целия обем бъбречна тъкансамо 25% функционират. Това не е достатъчно, за да се премахнат вредните токсини, уреята, поради което концентрацията им нараства. Клиничната картина на бъбречната недостатъчност е както следва:
- уринирането е нарушено. Вместо предписаните 800 ml дневно, пациентът отделя по-малко от 50 ml урина;
- здравето ви се влошава, появяват се слабост и умора;
- храносмилането е нарушено, апетитът изчезва, появяват се гадене и повръщане, сухота в устата е придружена от неприятна миризма;
- кожата става бледа, суха и постоянно сърби;
- сърдечно-съдовата система страда, причинявайки аритмия, повишено кръвно налягане и възможно разширяване на сърдечния мускул;
- мозък под въздействието на висока концентрация пикочна киселинасе уврежда, появяват се безсъние и увреждане на паметта, раздразнителност, намалени умствени способности;
- намаляването на хемоглобина и червените кръвни клетки води до анемия.
Увреждане на черния дроб
Системната амилоидоза често се проявява като увреждане на черния дроб. Амилоидните отлагания оказват натиск върху жлъчните пътища, кръвоносните съдове и чернодробните клетки, което води до нарушена функция на органите. При разграничаване на синдромите на амилоидозата те показват увеличен черен дроб, усетен при палпация.
Повърхността на черния дроб остава гладка, няма болка. При дълъг ход на заболяването рядко се развива чернодробна недостатъчност, която е свързана с регенеративните способности на органа.
Чернодробната амилоидоза се проявява със симптоми:
- Увеличен размер на черния дроб.
- Портална хипертония. Обикновено кръвта от вътрешните органи навлиза в черния дроб, където се пречиства и след това се връща обратно в кръвния поток. Когато чернодробните съдове се компресират от амилоид, налягането във вените на вътрешните органи се повишава. В резултат на това се появяват отоци на краката, диария с кръв и кървене в стомашно-чревния тракт.
- Жълтеница се появява рядко, само когато жлъчните пътища са притиснати от амилоидни отлагания. Ако това е причината, жълтеницата ще бъде придружена от сърбеж по кожата.
Сърдечна недостатъчност
Сърдечната амилоидоза се развива в първични и други наследствени форми. В резултат на амилоидни отлагания в миокарда и мембраните на сърцето кръвообращението се нарушава, мускулни клеткиумират.
Симптоми на заболяването:
- аритмия;
- рестриктивна кардиомиопатия;
- сърдечна недостатъчност.
Аритмията възниква на фона на амилоидни отлагания в сърдечния мускул, което нарушава проводимостта на нервните импулси. В резултат на това камерите на сърцето се свиват неравномерно и се появява аритмия. Болният се чувства замаян и припада. Поради нарушено кръвоснабдяване на мозъка е възможна смърт.
Рестриктивната кардиомиопатия възниква на фона на амилоидни отлагания в миокарда. В резултат на това сърдечният мускул се удебелява и става по-малко разтеглив, което води до лошо функциониране на сърдечните камери. Клиничната картина на заболяването е умора, задух, рязко понижаване на кръвното налягане по време на смяна хоризонтално положениедо вертикално положение.
При сърдечна недостатъчност кръвообращението в тялото е нарушено. Това се проявява чрез подуване и задух. Сърдечната недостатъчност, дължаща се на амилоидоза, не отговаря на стандартното лечение на сърдечно-съдови заболявания. Заболяването прогресира бързо и води до смърт в рамките на няколко месеца.
Увреждане на надбъбречните жлези и далака
 Надбъбречните жлези са жлези, разположени на всеки бъбрек и са отговорни за секрецията на хормони. Амилоидозата нарушава функцията на органа, като спира синтеза на хормони. Ако амилоидът се отложи в далака, органът се увеличава по размер, което се забелязва при палпация.
Надбъбречните жлези са жлези, разположени на всеки бъбрек и са отговорни за секрецията на хормони. Амилоидозата нарушава функцията на органа, като спира синтеза на хормони. Ако амилоидът се отложи в далака, органът се увеличава по размер, което се забелязва при палпация.
Обикновено далакът премахва деформирани клетки от кръвния поток, които се забиват в неговата структура. Амилоидните отлагания причиняват засядане на здрави червени кръвни клетки, тромбоцити и бели кръвни клетки в далака.
В резултат на това се развива анемия (обща слабост, бледа кожа, задух), тромбоцитопения (кървене от носа, кожни кръвоизливи), левкопения (податливост на инфекции).
Стомашно-чревни лезии
Чревната амилоидоза може да бъде генерализирана, когато абсорбцията на хранителни вещества е нарушена, и локална, когато амилоидните натрупвания имитират тумор. В първия случай се появяват симптоми като диария, загуба на тегло, слабост, психични разстройства и анемия. Във втория случай заболяването се характеризира със запек, коремна болка и подуване на корема.
Увреждане на ставите и мускулите
Амилоидът удря първи малки ставипо краката, ръцете, а с напредването на болестта се установява в лактите и коленете. Заболяването се характеризира с болка при движение, подуване на тъканта и зачервяване на кожата, повишена температура в засегнатата област и дисфункция на ставата.
Амилоидът се отлага незабелязано дълго време в съединителната тъкан, без да нарушава структурата на мускулите и без да се проявява. С течение на времето мускулните клетки се компресират, кръвоснабдяването им се нарушава и те умират. Заболяването се характеризира с мускулна слабост, болка, стягане и мускулна хипертрофия.
Диагностика на амилоидоза
Лекари с различни специализации - ревматолози, кардиолози и уролози, невролози, дерматолози и др., Могат да подозират диагноза като амилоидоза.Следователно диагнозата амилоидоза трябва да се основава на цялостна оценка на медицинската история, клинични признаци, лаборатория и инструментално изследване. За да се изследва състоянието на органите, се предписва ЕКГ, рентгеново изследване на хранопровода, ендоскопия и сигмоидоскопия. Ако се подозира бъбречна амилоидоза, диагнозата задължително включва ултразвук на коремната кухина.
Лечение на амилоидоза
Въпреки че има различни тежки заболяванияамилоидозата носи лоша прогноза. Факт е, че заболяването не може да бъде открито в ранните етапи и клиничните му прояви се забелязват много години след началото на заболяването. При диагноза като бъбречна амилоидоза лечението е само поддържащо, тъй като терапевтичните мерки не са ефективни.
При първото подозрение за наличието на заболяването е необходима хоспитализация в нефрологията за изследване пикочно-половата система, тъй като увреждането на бъбреците се счита за най опасна проява. Включват се и други специалисти, които изследват наличието на увреждане на други органи.
Ако диагнозата не разкрие сериозни нарушения във функционирането на жизненоважни важни органи, лечението на амилоидозата може да се проведе у дома, където пациентът трябва стриктно да спазва всички инструкции на лекаря. Лечението може да включва лекарства, диета, диализа и трансплантация на органи.
„Амилоидоза” е термин, който обединява група заболявания, които са много разнообразни клинични проявленияи се характеризират с извънклетъчно отлагане на неразтворими патологични фибриларни протеини в органи и тъкани. Тази патология е описана за първи път през 17 век. Костно-сагов далак при пациент с чернодробен абсцес. В средата на 19в. Вирхов използва ботаническия термин "амилоид" (от гръцки amylon - нишесте), за да опише извънклетъчния материал, открит в черния дроб при аутопсия, тъй като вярва, че той е подобен по структура на нишестето. Впоследствие се установява протеиновата природа на отлаганията, но терминът „амилоид“ се запазва и до днес.
През 20-те години. През 20-ти век Бенхолд предлага оцветяване на амилоид с конго червено, след което е открит ефектът на двойно пречупване в поляризирана светлина - промяна на тухленочервения цвят до ябълковозелен. През 1959 г. Коен и Калкинс използват електронна микроскопия, за да установят фибриларната структура на амилоида.
Клиничните идеи за амилоидозата също претърпяват еволюция: Рокитански през 1842 г. установява връзка между „мастната болест“ и туберкулозата, сифилиса и рикетсиозата; Wilkes през 1856 г. описва "мастни органи" при пациент, който няма съпътстващи заболявания; Аткинсън открива амилоидоза при пациенти с мултиплен миелом през 1937 г. Идентифицирани са сенилни (Soyka, 1876) и наследствени (Andrade, 1952) форми на заболяването, амилоидозата е разделена на генетична, първична и вторична и накрая през 1993 г. е приета класификацията на СЗО, базирана на спецификата на главния фибрилар. протеин на амилоид.
В нашата страна Е. М. Тареев, И. Е. Тареева, В. В. Серов имат голям принос за развитието на идеите за амилоидоза. Огромна роля в изучаването на първични и генетични варианти на амилоидоза и периодично заболяване принадлежи на О. М. Виноградова, чиито монографии, публикувани през 1973 и 1980 г., не са загубили своята актуалност днес.
В момента амилоидозата е клинично разделена на системна и локални форми. Сред системните форми, в зависимост от състава на фибриларните отлагания, се разграничават четири вида ( ).
Местните форми на амилоидоза понастоящем включват болестта на Алцхаймер (А-бета, фибрилите се състоят от β-протеин, отложен в мозъка), амилоидоза на панкреатичните острови, вероятно имаща патогенетична връзка с диабет тип 2, амилоидоза, възникваща при ендокринни тумори, амилоидни тумори на кожата, назофарингеалната област, пикочния мехур и други редки видове.
AL амилоидоза
Развитието на AL амилоидоза е възможно при миелом, болест на Waldenström, В-клетъчни лимфомии може да бъде идиопатичен при първична амилоидоза. Всички тези опции са комбинирани обща патогенеза, най-трудно се разпознава първичната амилоидоза поради липсата очевидни признаци хематологично заболяване, затова си струва да се спрем подробно на тази форма.
При първична амилоидоза, доброкачествена дискразия на плазмени клетки, свързана множествена миелома, анормални клонове на плазмени клетки от костен мозък произвеждат амилоидогенни имуноглобулини. Някои аминокиселини в вариабилните области на леките вериги на тези имуноглобулини заемат необичайна позиция, което води до тяхната нестабилност и предизвиква тенденция към фибрилогенеза. При пациенти с първична амилоидоза съдържанието на плазмени клетки в костния мозък се повишава до 5-10% (обикновено по-малко от 4%, при миелома - повече от 12%) и те произвеждат определен изотип на имуноглобулиновите леки вериги, които преобладава в имунохистохимичното оцветяване. В кръвта и урината се откриват свободни моноклонални леки вериги от преобладаващия ламбда или (по-рядко) капа изотип, но тяхното съдържание е по-ниско, отколкото при мултиплен миелом.
Клиничната картина на първичната амилоидоза е разнообразна и се определя от преобладаващото засягане на определени органи в патологичния процес - сърце, бъбреци, нервна система, стомашно-чревен тракт, черен дроб и др. Първите симптоми са слабост и загуба на тегло, но при това етап, преди появата на органни симптоми, диагнозата се поставя изключително рядко.
Прицелните органи за AL амилоидоза най-често са бъбреците и сърцето. Увреждането на бъбреците се проявява с нефротичен синдром, който продължава дори с появата на хронична бъбречна недостатъчност; хематурия и артериална хипертония не са типични.
Когато амилоидът се отложи в миокарда, различни нарушенияритъм, прогресивна сърдечна недостатъчност, която може да бъде предшествана от асимптоматични промени в ЕКГ под формата на намаляване на напрежението на вълната. Ехокардиографското изследване разкрива концентрично удебеляване на стените на лявата и дясната камера, намаляване на обема на сърдечните кухини, умерено намаляване на фракцията на изтласкване и диастолна дисфункция на миокарда на лявата камера.
Често се наблюдават симптоми на засягане на нервната система – вегетативна, под формата ортостатична хипотония, и периферна - под формата на нарушения на чувствителността. През последните години започнаха да се описват и лезии на централната нервна система, въпреки че преди това се смяташе, че те не са характерни за първичната амилоидоза.
Диспептичните симптоми (усещане за пълнота, запек, диария) и синдромът на малабсорбция могат да бъдат причинени както от увреждане на автономната нервна система, така и от амилоидоза на стомашно-чревния тракт. Много характерна е хепатомегалията, чиято природа трябва да се разграничава между конгестия поради сърдечна недостатъчност и амилоидно увреждане на черния дроб. Последното се потвърждава от повишаване на нивото на алкалната фосфатаза в кръвния серум. Далакът често е засегнат, но спленомегалията не винаги се открива и е голяма клинично значениене притежава.
Макроглосията, класически признак на първична амилоидоза, се наблюдава при 20% от пациентите; инфилтрацията на меките тъкани може да доведе до атрофия на мускулите, кожата, дистрофия на ноктите, алопеция и появата на туморни образувания - амилоид.
По-рядко се срещат съдови лезии, симптомите на които са периорбитална пурпура - „очи на миеща мечка“ и екхимози. Може да възникне кървене, включително кървене от пикочния мехур, причинено както от промени в съдовата стена, така и от нарушение на системата за кръвосъсирване, предимно от дефицит на X фактора, който се свързва с амилоида. Тромбоцитозата, характерна за амилоидозата, също често се обяснява с дефицит на коагулационни фактори.
Белодробната амилоидоза често се открива само при аутопсия. Въпреки това, в някои случаи задухът, хемоптизата и хидротораксът могат да бъдат причинени не само от застойна сърдечна недостатъчност и нефротичен синдром, но и от амилоидно белодробно заболяване. Възможно е отлагане на амилоид в алвеолите и развитие на белодробен амилоид. Рентгеновите лъчи могат да разкрият ретикуларни и нодуларни промени в белодробната тъкан.
Увреждането на надбъбречните жлези може да доведе до надбъбречна недостатъчност, която често остава неразпозната, тъй като хипотонията и хипонатремията се считат за симптоми на сърдечна недостатъчност и увреждане на автономната нервна система. При 10-20% от пациентите хипотиреоидизмът може да се появи като проява на увреждане на щитовидната жлеза, често се среща увеличение на субмандибуларните слюнчени жлези.
Диагнозата първична амилоидоза, в допълнение към посочените клинични характеристики, които могат да бъдат сходни при вторичната амилоидоза, се основава на редица лабораторни данни. При 85% от пациентите имуноелектрофореза на протеини в серума и урината разкрива моноклонални имуноглобулини. При рутинни изследвания същите моноклонални имуноглобулини се откриват в урината под формата на протеин на Bence Jones. Биопсия на костен мозък позволява диференциална диагнозас множествена миелома и също така разкриват умерено увеличение на броя на плазмените клетки и тяхната моноклоналност с имунохистохимично оцветяване.
Въпреки това, дори комбинацията от характерна клинична картина и наличието на моноклонални плазмени клетки и протеини все още не е достатъчна за потвърждаване на диагнозата първична амилоидоза. Тук решаваща роля играят данните от биопсията. Най-малко инвазивна е аспирацията на подкожната мастна тъкан на предната коремна стена, давайки 80-90% положителни резултатиза AL амилоидоза (този метод все още не е намерил приложение у нас). Биопсията на венците и ректалната лигавица има определена диагностична стойност, но процентът на положителните резултати варира в широки граници, в зависимост от стадия на процеса, затова е препоръчително да се направи биопсия на един от засегнатите органи - бъбрек, черен дроб , сърце, което дава почти 100% положителни резултати за амилоидоза AL-тип.
Първо, биопсичният материал се оцветява с конго червено. Ако се открие конгофилия на изследвания материал, той трябва да се изследва в поляризирана светлина; ефектът на двойно пречупване е характерен само за амилоид; други конгофилни вещества не придобиват ябълково-зелен цвят. След това е желателно типизиране на амилоид. Най-точен е имунохистохимичният метод, използващ моноклонални антитела към амилоидните прекурсорни протеини. В момента обаче той практически не се предлага у нас. Ето защо за диагностика се използва оцветяване с разтвори на алкален гуанидин или калиев перманганат, което позволява, макар и индиректно, да се определи вида на фибриларните отлагания.
Прогнозата за първична амилоидоза е по-лоша, отколкото за други форми на заболяването, средна продължителностживотът не надвишава две години; при наличие на сърдечно увреждане или мултисистемно увреждане без лечение пациентите умират в рамките на няколко месеца. Повечето често срещани причинисмъртните случаи са сърдечна и бъбречна недостатъчност, сепсис, съдови усложнения и кахексия. Патогенетичното сходство с множествения миелом ни позволява да очакваме инхибиране на прогресията на заболяването с химиотерапия, прилагана за потискане на моноклоналните плазмени клетки. Има няколко схеми на лечение ().
Използването на химиотерапия, ако лечението е успешно, може да увеличи продължителността на живота на пациентите с 10 до 18 месеца. Но ефективността на терапията е ниска, по-специално поради факта, че в много случаи прогресията на заболяването води до смърт на пациентите преди завършване на курса на лечение, както и поради развитието на цитопения, инфекциозни усложнения, фатални аритмии по време на лечение с ултрависоки дози дексазон. Използването на високи дози мелфолан с автоложна трансплантация на стволови клетки позволява постигане на ремисия в повече от 50% от случаите, но използването на този метод е ограничено от тежестта на състоянието, възрастта на пациентите, функционални нарушенияот сърцето и бъбреците. В много случаи е възможна само симптоматична поддържаща терапия.
АА амилоидоза
Развитието на АА амилоидоза възниква по време на хронични възпалителни процеси, прекурсорите на АА амилоида са серумни острофазови протеини, α-глобулини, произведени от клетките различни видове, главно неутрофили и фибробласти. Вторичната амилоидоза се развива с ревматоиден артрит, анкилозиращ спондилит, псориатичен артрит, различни тумори, лимфогрануломатоза, улцерозен колит и болест на Crohn, с периодично заболяване (фамилна средиземноморска треска), както и с туберкулоза, остеомиелит, бронхиектазии.
Характерни клинични характеристики на АА амилоидозата са бъбречно увреждане при повечето пациенти, както и относително рядко увреждане на черния дроб и/или далака (около 10%) и сърцето (открива се само чрез ехокардиография). Макроглосията не е характерна за вторичната амилоидоза. Диагнозата се основава на комбинация от бъбречна амилоидоза и хронично възпалително заболяване, потвърдено чрез имунохистохимично оцветяване на биопсичен материал, у нас се използват индиректните методи за оцветяване, споменати по-горе.
Прогнозата до голяма степен зависи от естеството на основното заболяване; по време на естествения ход една трета от пациентите развиват бъбречна недостатъчност 5 години от момента на откриване на протеинурия. При периодично заболяване петгодишната преживяемост е 25%.
Лечението се основава на потискане на фокуса - източникът на производство на серумни прекурсорни протеини. Отстраняването на тумори, секвестректомия, резекция на червата, лечение на туберкулоза, намаляване на активността на ревматоиден артрит (с използване на цитостатици) водят до спиране на прогресията на амилоидозата, а понякога и до обратното развитие на клиничните прояви, по-специално нефротични синдром.
Употребата на колхицин при периодично заболяване е метод на избор, неговата ефективност е доказана, лечението предотвратява развитието на амилоидоза и инхибира нейното прогресиране. При други форми на вторична амилоидоза ефективността на колхицин не е потвърдена.
Сенилни и наследствени форми на системна амилоидоза, както и локални форми, са редки; диализната амилоидоза е добре известна на специалистите и практически не се среща в общата практика.
Симптоматичната терапия зависи не от вида на амилоидозата, а от засегнатите целеви органи ( ).
Амилоидозата, особено първичната, се счита за необичайна патология, но в действителност не е толкова рядка, колкото е трудна за диагностициране. Адекватната диагноза изисква не само познаване на клиничната картина и патогенезата на заболяването, но и наличието на определени диагностични възможности. За да илюстрираме тази точка, представяме нашите собствени данни ( ). В отделението по нефрология на Московската градска клинична болница S.P. Botkin през 1993-2003 г. Наблюдавани са 88 пациенти с диагностицирана амилоидоза.
Диагнозата е потвърдена морфологично при всички пациенти с АЛ амилоидоза, сенилна и неуточнена амилоидоза, а при 30 пациенти с вторична амилоидоза - общо 53 случая. При 12 пациенти е извършена бъбречна биопсия, при двама пациенти е извършена чернодробна биопсия, при 8 пациенти е извършена чревна биопсия, при 12 е извършена биопсия на венците, а при други 19 диагнозата е потвърдена с морфологично изследване на срезови материал.
В повечето случаи диагнозата амилоидоза се поставя за първи път в резултат на преглед в нефрологичното отделение. Направихме сравнение между пациенти с AL амилоидоза на референтни и клинични диагнози ( ).
Само в два от 20 случая (10%) насочената диагноза е „първична амилоидоза“, като единият от тези пациенти е диагностициран в Клиниката по терапия и професионални болести на ВМА, а другият в чуждестранна клиника.
Всички пациенти, които са били диагностицирани с миелом с развитието на AL амилоидоза, са прехвърлени в хематологичните отделения. От 11 пациенти с първична амилоидоза, седем пациенти са получили химиотерапия с комбинация от мелфолан и перорален преднизолон в интермитентни курсове, четирима от тях - в комбинация с диализно лечение, а друг пациент - само диализа и симптоматично лечение. От тези пациенти пет души са починали в рамките на период от две седмици до две години от началото на лечението (всички с бъбречна недостатъчност и множество органни увреждания), един пациент е на диализа, един пациент е насочен за автоложна трансплантация на стволови клетки и един пациентът получава лечение до сегашно време. При един пациент химиотерапията е отложена поради наличие на продължителна стомашна язва, а други двама пациенти са отказали лечение.
Сред пациентите с вторична амилоидоза в нашето проучване преобладават пациентите с ревматоиден артрит; хроничният остеомиелит и псориатичният артрит са на второ място сред причините; други заболявания са по-рядко срещани ( ).
Лечението на ревматоиден артрит и псориатичен артрит се провежда с цитостатици (метатрексат, азатиоприн), въпреки че в много случаи възможностите за лечение са ограничени поради наличието на хронична бъбречна недостатъчност и съпътстваща патология. Пациентите с хроничен остеомиелит са изпратени в отделения по гнойна хирургия. Пациентите с анкилозиращ спондилит и болестта на Крон получиха специфично лечение, пациентите с ХОББ и туберкулоза също бяха изпратени в специализирани болници. Един от пациентите с тумор на стомаха беше успешно опериран и в продължение на четири години наблюдение нефротичният синдром постепенно регресира; в други случаи на тумори разпространението на процеса позволява само симптоматична терапия; пациентът с лимфогрануломатоза е приет да се крайно състояние. Смъртността сред пациентите с вторична амилоидоза е 38% (поради пациенти с напреднали лезии по време на диагнозата). Всички пациенти с периодично заболяване са получили терапия с колхицин.
Характеристиките на диагностиката и прилагането на съвременни методи за лечение на първична амилоидоза могат да бъдат илюстрирани със следния пример: пациент К., на 46 години, е хоспитализиран за първи път в края на октомври 2002 г. с оплаквания от подуване на краката, сърцебиене и аменорея. В анамнезата - настинки, апендектомия, две нормални спешни раждания, без признаци на бъбречно заболяване, без хронични заболявания. През април 2002 г. тя се мести остра пневмонияв горния лоб на десния бял дроб, лекуван е амбулаторно, получава инжекции абактал и линкомицин. Поради локализацията на пневмония е прегледана в клиника за туберкулоза, като диагнозата туберкулоза е изключена. В началото на юни за първи път се появяват отоци по краката, за които не е била на преглед. Подуване чрез кратко времебяха ликвидирани сами, след което възобновени. Пациентът е хоспитализиран в терапевтична болница; изследването показва протеинурия до 1,65%, хипопротеинемия (общ серумен протеин 52 g / l), нормално кръвно налягане (120/80 mm Hg), непроменен седимент в урината, плазмените нива на креатинин също са в рамките на нормата граници. Поставена е диагноза остър гломерулонефрит, проведено е лечение с ампицилин, камбанка, хепарин, триампур и е направена тонзилектомия. Протеинурията продължава, отокът постепенно се увеличава и следователно за по-нататъшно изследване и лечение пациентът с диагноза хроничен гломерулонефрит е изпратен в болницата на името на. С. П. Боткина.
При преглед кожата е чиста, с нормален цвят, анасарка, масивен, плътен оток, открива се асцит, периферните лимфни възли не са увеличени. Кръвно налягане 110/70 mm Hg. Чл., Сърдечните звуци са звучни, ясни, ритмични, сърдечна честота 90 удара / мин, черният дроб и далакът не са увеличени, диурезата е до 1000 ml / ден, изпражненията са редовни, без патологични примеси. При изследването се установява нефротичен синдром - протеинурия 3 g/l, оскъден седимент в урината, хиподиспротеинемия, хиперлипидемия (общ серумен протеин 39 g/l, албумин 12 g/l, глобулини 7-30-15-19%, съответно α 1 -α 2 -β-γ холестерол 17,8 mmol/l, β-липопротеини 250 IU), при анализ на урината за протеин на Bence-Jones - реакцията е отрицателна, дневната екскреция на 17-KS не се намалява. Клиничният кръвен тест и други биохимични параметри са в нормални граници, коагулограмата показва тежка хиперфибриногенемия, повишено ниво на RKFM. Изследване на кръвни имуноглобулини: Ig-A - 0,35, Ig-M - 35,7 (две норми), Ig-G - 1,96 g/l. Рентгенография на гръдни органи, череп и тазови кости, ултразвук на коремна кухина, бъбреци, щитовидна жлеза, ECHO-CG без патология, ултразвук на таза - признаци на аденомиоза на тялото на матката, ендоскопия - рефлуксен езофагит, хроничен гастрит. При преглед от невролог не е открита патология, онкологът диагностицира фиброкистозна мастопатия.
За изясняване на генезиса на нефротичния синдром е извършена тънкоиглена пункционна биопсия на десния бъбрек под локална анестезия и ехографски контрол, без усложнения. При изследване на биопсичен препарат се забелязва отлагане на амилоид в гломерулния мезангиум и в екстрагломерулните съдове. Амилоидът натоварва до 25% от гломерулните съдови бримки. Имунохистохимично изследване не открива специфична луминесценция. При третиране на препаратите с алкален разтвор на гуанидин в продължение на 2 часа конгофилите и свойствата им в поляризирана светлина се запазват, което е характерно за AL амилоидозата.
За изясняване същността на АЛ амилоидозата в лаборатория Имунотест е проведено имунохимично изследване на кръв и урина. Открита е M-ламбда парапротеинемия с намаляване на нивото на поликлоналните имуноглобулини и парапротеинурия от типа на Bence-Jones на фона на масивна неселективна протеинурия. Пациентът е консултиран от хематолог, предполага се наличието на болест на Waldenström и е направена биопсия на костен мозък. Заключение: в съществуващите кухини на костния мозък се виждат клетки от трите зародиша на нормалната хематопоеза, както и лимфоидни клетки, които не образуват клъстери. Диагнозата болест на Waldenström беше отхвърлена поради липсата на лимфоидна инфилтрация на костния мозък, увеличение на лимфните възли и далака и липсата на туморен субстрат.
Установена е диагноза първична амилоидоза с увреждане на бъбреците, нефротичен синдром, запазена бъбречна функция, не са открити признаци на увреждане на други органи. През януари 2003 г. започва химиотерапия с мелфолан 16 mg/ден и преднизолон 100 mg/ден, курсове от четири дни на всеки шест седмици. Провежда се и симптоматично лечение: фуроземид, верошпирон, калиеви препарати, фамотидин, трансфузии на албумин. Към днешна дата са проведени пет курса химиотерапия с добра поносимост, отокът е намалял, протеинурията е намаляла до 1,8 g/l и тежестта на хиподиспротеинемията леко е намаляла (общ протеин 46 g/l, албумин 18 g/l, α 2 -глобулини 20%). Бъбречната функция остава ненарушена, плазменият креатинин е 1,3 mg/dL и не са открити признаци на увреждане на други органи и системи по време на динамичните контролни изследвания.
Този случай ясно илюстрира факта, че за диагностицирането на амилоидоза е необходимо морфологично, имунологично и имунохимично изследване. Така при нашия пациент най-очевидната клинична диагноза беше „хроничен гломерулонефрит“ и при липса на възможност за извършване на бъбречна биопсия тази диагноза най-вероятно би била поставена. Няма клинични признаци за системен характер на заболяването, хронично възпалителен процес, пациентът не е имал заболяване на кръвоносната система, с изключение на повишаване на нивото на Ig-M. И само данните, получени от изследването на бъбречната биопсия, включват биопсия на костен мозък и имунохимично изследване, което заедно прави възможно диагностицирането на първична амилоидоза преди появата на системно увреждане. Патогенетична терапиязапочна, макар и на фона на вече развит нефротичен синдром, но преди появата на бъбречна недостатъчност и когато само 25% от гломерулите са натоварени с амилоид, което има относително благоприятна прогноза.
В заключение отбелязваме, че амилоидозата е сериозно заболяванес високо нивосмъртност, която е изключително трудна за диагностициране, но навременният и качествен преглед на пациентите позволява да се постави диагнозата по-рано, и навременна срещаадекватната терапия от своя страна дава възможност за подобряване на прогнозата при тази група пациенти.
Литература
- Varshavsky V. A., Proskurneva E. P. Значението и методите на морфологичната диагностика на амилоидозата при съвременна медицина// Практическа нефрология. - 1998. - 2:16-23.
- Виноградова О. М. Първични и генетични варианти на амилоидоза. - М.: Медицина, 1980.
- Захарова Е. В., Хрикина А. В., Проскурнева Е. П., Варшавски В. А. Случай на първична амилоидоза: трудности при диагностицирането и лечението // Нефрология и диализа. - 2002. - 1:54-61.
- Рамеев В. В. Характеристики на увреждане на бъбреците при AA и AL амилоидоза: дис... канд. пчелен мед. Sci. - М., 2003.
- Козловская Л. В., Варшавски В. А., Чегаева Т. В. и др. Амилоидоза: модерен видкъм проблема // Практическа нефрология. - 1998. - 2:24-26.
- Rodney H., Raymond L.C. и Skinner M. // Системните амилоидози; New England Journal of Medicine, 1997. - 337:898-909.
- Dhodapkar M.V., Jagannath S., Vesole d. et al // Лечение на AL-амилоидоза с дексаметазон плюс алфа интерферон / Leuc Lymphoma. 1997. - 27 (3-4): 351-365
- Gertz M.A., Lacy M.Q., Lust J.A. et all // Изпитване фаза II на висока доза дексаметазон за предварително лекувана имуноглобулинова амилоидоза с лека верига. Am J Hematol 1999,61(2):115-119.
- Gertz M.A., Lacy M.,Q., Lust J.A. et al // Фаза II проучване на висока доза дексаметазон при нелекувани пациенти с първична системна амилоидоза. Med Oncol 1999.- 16(2):104-109
- Sezer O., Schmid P., Shweigert M. et al // Бързо обръщане на нефротичния синдром, дължащо се на първична системна AL амилоидоза след VAD и последваща химиотерапия с високи дози с автоложна подкрепа за продажба на стволови. Трансплантация на костен мозък. 1999. - 23 (9): 967-969.
- Sezer O., Neimoller K., Jakob C. et al // Нови подходи към лечението на първична амилоидоза. Експертно мнение Разследване на неприятности. 2000. - 9 (10): 2343-2350
- Sezer O., Eucker J., Jakob C., Possinger K. // Диагностика и лечение на AL амилоидоза. Клин Нефрол. 2000. - 53 (6): 417-423.
- Skinner M. "Амилоидоза" Текуща терапия в алергията, имунологията и ревматологията. Мосби-годишна книга. 1996. - 235-240.
- Паладини Г., Анеси Е., Перфети В. и др. Модифициран режим на дексаметазон с високи дози за първична системна (AL) амилоидоза. Британско списание по хематология. 2001. - 113: 1044-1046.
Е. В. Захарова
град Москва клинична болницатях. С. П. Боткина
Таблица 2. Схеми на лечение на първична амилоидоза
- Циклично перорално приложение на мелфолан (0,15-0,25 mg/kg телесно тегло на ден) и преднизолон (1,5-2,0 mg/kg на ден) за четири до седем дни на всеки четири до шест седмици за една година, до достигане на курсовата доза от 600 мг
- Перорално приложениемелфолан в доза от 4 mg/ден в продължение на три седмици, след това след двуседмична пауза - 2-4 mg/ден четири дни в седмицата непрекъснато до достигане на курсовата доза от 600 mg, в комбинация с преднизон
- Интравенозно приложение на високи дози мелфолан (100-200 mg/m² телесна повърхност за два дни), последвано от автоложна трансплантация на стволови клетки
- Интравенозно дексаметазон 40 mg за четири дни на всеки три седмици - осем цикъла
- Интравенозно приложение на дексаметазон в доза от 40 mg на първия-четвъртия, 9-12-ия и 17-20-ия ден от 35-дневния цикъл, три до шест цикъла, последвано от употребата на а-интерферон в доза от 3- 6 милиона единици три пъти на ден седмично
- Схема винкристин-доксорибуцин-дексаметазон (VAD).
