آمیلوئیدوز سیستمیک: تشخیص، تشخیص افتراقی، درمان. سیر بالینی و تظاهرات اصلی آن
آمیلوئیدوز- بیماری که با اختلال متابولیک مشخص می شود، در نتیجه یک ماده جدید برای بدن (آمیلوئید) تشکیل می شود که در اندام ها رسوب می کند و عملکرد آنها را مختل می کند.
آمیلوئید یک گلیکوپروتئین پیچیده است که در آن پروتئین های فیبریلار و کروی ارتباط نزدیکی با پلی ساکاریدها دارند. فیبریل آمیلوئید از پروتئین های پلی پپتیدی تشکیل شده است. علاوه بر پروتئین فیبریلار، آمیلوئید حاوی پروتئین دیگری نیز می باشد - به اصطلاح P-component که برای همه انواع آمیلوئید یکسان است. اعتقاد بر این است که جزء P یک پروتئین طبیعی سرم مرتبط با فیبریل های آمیلوئید است.
آمیلوئیدوز می تواند به عنوان عارضه هر بیماری رخ دهد یا به عنوان یک فرآیند مستقل ایجاد شود.
در حال حاضر، بسته به علت، انواع مختلفی از آمیلوئیدوز وجود دارد که ترکیب بیوشیمیایی خود را از فیبرهای آمیلوئید دارند.
آمیلوئیدوز اولیه (ایدیوپاتیک) بدون ایجاد می شود دلایل قابل مشاهدهو اندام های مختلف (قلب، کلیه ها، روده ها، کبد، سیستم عصبی) را تحت تاثیر قرار می دهد. شکل بیوشیمیایی آمیلوئیدوز اولیه شکل AL است، پیش ساز این آمیلوئید Ig و زنجیره سبک ایمونوگلوبولین است. با توجه به ساختار آمیلوئید و ماهیت ضایعه اعضای داخلیدوز اولیه آمیلوئید (ایدیوپاتیک) نزدیک به آمیلوئیدوز در مولتیپل میلوما است که در حال حاضر به یک گروه جداگانه تقسیم شده است.
آمیلوئیدوز ارثی (ژنتیکی) با یک ضایعه کلیوی غالب، ترکیبی از آسیب کلیه و سیستم عصبی. در کشور ما آمیلوئیدوز ارثی معمولاً با یک بیماری دوره ای همراه است که به صورت اتوزومال غالب منتقل می شود. در این بیماری، آمیلوئیدوز ممکن است تنها تظاهرات باشد. شکل بیوشیمیایی آمیلوئیدوز ارثی AF است (پیش ساز آمیلوئید پرآلبومین است). در مورد بیماری دوره ای، فرم بیوشیمیایی AA است (پیش ساز پروتئین SAA است).
آمیلوئیدوز اکتسابی (ثانویه) اغلب رخ می دهد و در آرتریت روماتوئید، بیماری Bechterew، سل، چرک مزمن - استئومیلیت، برونشکتازی، آبسه مزمن ریه، کمتر در کولیت اولسراتیو، پسوریازیس، لنفوگرانولولوماتوز ثانویه کلیوی، تومورهای شیمیایی بیولوژیکی، سیوفولوماتوز کلیوی و غیره ایجاد می شود. - AA (پیش ساز سرمی آن پروتئین SAA است که توسط سلول های کبدی سنتز می شود).
آمیلوئیدوز پیری نتیجه اختلالات متابولیسم پروتئین است که در مغز، پانکراس و قلب یافت می شود. فرمول بیوشیمیایی AS است (پیشآلبومین است).
آمیلوئیدوز موضعی بدون دلیل ظاهری ایجاد می شود، فرمول بیوشیمیایی آن AE است (پیش ساز ناشناخته است).
پاتوژنز.تنها پیوندهای مجزای پاتوژنز به خوبی شناخته شده است (طرح 22). از نمودار بر می آید که تحت تأثیر جهش ژنی و همچنین قرار گرفتن در معرض عوامل خارجیتغییرات ایمنی - کاهش می یابد
تعداد لنفوسیت های T این منجر به کاهش اثر کنترلی آنها بر روی سیستم B لنفوسیت ها می شود. در نتیجه، تعداد سلول های B حامل ایمونوگلوبولین های طبیعیو تعداد سلول های B که پیش سازهای فیبریل آمیلوئید را سنتز می کنند افزایش می یابد. آمیلوئیدوبلاست ها در مقدار زیاد پروتئین فیبریلار تولید می کنند که باعث سنتز آمیلوئید در مقادیر زیاد می شود.
اما به دلیل نقص ژنتیکی آمیلوئیدوکلاست ها که به کاهش فعالیت آنزیمی آنها کمک می کند، جذب آمیلوئید کافی رخ نمی دهد. در نتیجه، افزایش رسوب آمیلوئید در بافت ها و اندام ها وجود دارد [Mukhin N.A.، 1981].
در مولتیپل میلوما، آمیلوئیدوز در نتیجه افزایش تولید پارپروتئین توسط سلول های پلاسما ایجاد می شود که برای ساخت آمیلوئید استفاده می شود. ترکیب آمیلوئید در اشکال مختلف آمیلوئیدوز متفاوت است که با ترکیب پروتئین فیبریل های آمیلوئید مشخص می شود.
با آسیب میوکارد، اعصاب محیطی(که عمدتاً در شکل ایدیوپاتیک آمیلوئیدوز مشاهده می شود) آمیلوئید در اطراف رشته های کلاژن رسوب می کند. بافت همبند. رسوب آمیلوئید در اطراف رشته های شبکه ای در ضایعات مشاهده می شود
کلیه ها، روده ها، کبد، غدد فوق کلیوی، پانکراس (با آمیلوئیدوز ارثی و ثانویه). با این حال، ترکیبی از رسوبات آمیلوئید پری کلاژن و پری رتیکولار امکان پذیر است که ضایعات ترکیبی اندام ها و سیستم های مختلف را ایجاد می کند.
با رسوب آمیلوئید در بافت ها، تعداد عناصر فعال، کاردیومیوسیت ها، هپاتوسیت ها، رشته های عصبی و گلومرول های کلیوی کاهش می یابد که متعاقباً منجر به ایجاد نارسایی اندام می شود.
بنابراین، در قلب، آمیلوئید در زیر اندوکارد، در استروما و عروق میوکارد و همچنین در امتداد وریدهای اپی کاردیوم رسوب می کند. در همان زمان، اندازه قلب به شدت افزایش می یابد و تعداد کاردیومیوسیت ها به سرعت کاهش می یابد. همه اینها منجر به کاهش می شود عملکرد انقباضیانفارکتوس میوکارد و نارسایی قلبی، و همچنین اختلالات هدایت و ریتم قلب. در مغز مبتلا به آمیلوئیدوز پیر، آمیلوئید در پلاکهای به اصطلاح پیری قشر، عروق و غشاها یافت میشود. در پوست، آمیلوئید در پاپیلاها و دیواره های عروقی رسوب می کند که منجر به آتروفی شدید اپیدرم می شود. در کبد، آمیلوئید بین رتیکولواندوتلیوسیت های ستاره ای عروق سینوسی، در دیواره عروق، مجاری و در بافت همبند مجاری پورتال رسوب می کند. با تجمع آمیلوئید، سلول های کبدی آتروفی می شوند.
در کلیه ها، آمیلوئید در غشای مویرگ های گلومرولی و لوله های نفرون، در مزانژیوم، حلقه های مویرگی و در امتداد شریان ها رسوب می کند. با تجمع آمیلوئید، بیشتر نفرونها آتروفی میشوند، میمیرند یا با بافت همبند جایگزین میشوند - یک کلیه کوچک شده با آمیلوئید ظاهر میشود. این فرآیند را می توان به صورت نمودار زیر نشان داد:
پروتئینوری -> سندرم نفروتیک -> نارسایی کلیه.
بر این اساس، سه مرحله در تصویر بالینی متمایز می شود: 1) اولیه (پروتئینوری). 2) مستقر شده (نفروتیک). 3) ترمینال (آزوتمیک).
تصویر بالینی.تظاهرات آمیلوئیدوز متنوع است و با موارد زیر مشخص می شود: 1) محلی سازی آمیلوئید در یک اندام خاص. 2) درجه شدت رسوبات آمیلوئید در اندام. 3) بیماری اصلی که آمیلوئید در برابر آن ایجاد شده است (با شکل ثانویه آمیلوئیدوز).
ممکن است مشکلاتی در تشخیص ایجاد شود، زیرا تظاهرات بالینی بیماری فقط با مقدار معینی از آمیلوئید رسوب شده قابل توجه خواهد بود. در این راستا، یک دوره "نهفته" از لحظه رسوب آمیلوئید تا ظهور علائم اختلال در عملکرد یک اندام یا سیستم اجتناب ناپذیر است.
تصویر بالینی به ویژه در صورت آسیب کلیوی روشن است که شایع ترین محل رسوبات آمیلوئید است.
در مرحله I جستجوی تشخیصی V مرحله اولیهعملاً هیچ اطلاعاتی که نشان دهنده آسیب کلیه در اثر آمیلوئیدوز باشد را نمی توان به دست آورد. شکایات بیماران با بیماری زمینه ای (با آمیلوئیدوز ثانویه) همراه است.
در تاریخچه اطلاعاتی در مورد وجود یک بیماری خاص (سل ریوی، استئومیلیت، آرتریت روماتوئید و غیره)، دوره آن و درمان وجود دارد. این اطلاعات به خودی خود اجازه تشخیص آمیلوئیدوز کلیه را نمی دهد، اما توجه پزشک را به این احتمال جلب می کند.
در مرحله پیشرفته آمیلوئیدوز، بیماران به دلیل ایجاد سندرم نفروتیک، از کاهش مقدار ادرار، ادم با شیوع و شدت متفاوت و همچنین شکایت از ضعف، بی اشتهایی و کاهش عملکرد شکایت دارند. همراه با آنها، در آمیلوئیدوز ثانویه، شکایاتی از تظاهر بیماری زمینه ای وجود دارد.
در مرحله پایانی، شکایات ناشی از ایجاد نارسایی مزمن کلیوی است: از دست دادن اشتها، حالت تهوع، استفراغ (اختلالات سوء هاضمه)، سردرد، اختلالات خواب (اختلالات سیستم عصبی)، خارش پوست.
در مرحله دوم جستجوی تشخیصی، فقط علائم مشخصه بیماری زمینه ای (با آمیلوئیدوز ثانویه) را می توان در مراحل اولیه تشخیص داد.
که در مرحله پیشرفتهآشکار می کند: 1) هیپوستازهای محلی سازی و بیان مختلف؛ با احتباس قابل توجه مایع در بدن، هیدروتوراکس، هیدروپریکارد، آسیت گذرا ممکن است ظاهر شود. 2) فشار خون شریانی (در 12-20٪ از بیماران مبتلا به آمیلوئیدوز رخ می دهد)، اتساع و هیپرتروفی بطن چپ. 3) افزایش کبد و طحال به دلیل رسوب آمیلوئید در بافت ها (کبد و طحال متراکم، بدون درد، با لبه نوک تیز هستند). 4) علائم بیماری زمینه ای (با آمیلوئیدوز ثانویه).
که در مرحله ترمینالعلائم با شدت نارسایی کلیه تعیین می شود: 1) سندرم دیستروفیک (تغییرات در پوست و غشاهای مخاطی). 2) سندرم سروزی مفصلی (استئوآرتروپاتی، نقرس ثانویه، پریکاردیت خشک، پلوریت). 3) فشار خون شریانی
در مرحله III جستجوی تشخیصی آمیلوئیدوز، مهم ترین اطلاعات برای تشخیص به دست می آید که می توان آنها را گروه بندی کرد. به روش زیر: 1) سندرم ادراری; 2) نقض متابولیسم پروتئین و چربی؛ 3) تشخیص رسوبات توده های آمیلوئید.
سندرم ادراری: 1) پروتئینوری - علامت اصلیآمیلوئیدوز، در تمام اشکال آن، اما اغلب در آمیلوئیدوز ثانویه ایجاد می شود. پروتئینوری معمولاً قابل توجه است، روزانه 2-20 گرم پروتئین آزاد می شود که قسمت اصلی آن آلبومین است. در مقادیر کمتر، گلوبولین ها دفع می شوند، ممکن است پیش ساز آمیلوئید سرم (پروتئین SAA) از طریق ادرار دفع شود. در مرحله پایانی، پروتئینوری ادامه دارد. در ادرار، گلیکوپروتئین های a- و به خصوص y- را می توان تشخیص داد.
با توجه به درجه پروتئینوری، گچ های هیالین و کمتر دانه ای یافت می شود. به ندرت، میکرو هماچوری یا لکوسیتوری تشخیص داده می شود، اما شدت آن با درجه پروتئینوری مطابقت ندارد (همانطور که با گلومرولونفریت مشاهده می شود). درجه اختلالات متابولیسم لیپید در آمیلوئیدوز مربوط به لیپویدوری با حضور کریستال های دوشکست کننده در رسوب ادرار است.
اختلالات متابولیسم پروتئین و لیپید: 1) هیپوپروتئینمی همراه با هیپوآلبومینمی و هیپر<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
برای تشخیص مهم است تشخیص توده های آمیلوئید در اندام ها و بافت ها:در کبد (50٪ موارد)، طحال (با بیوپسی سوراخ)، غشای مخاطی لثه و رکتوم.
در مرحله اولیه (پروتئینوری)، بیوپسی از غشای مخاطی لثه اغلب نتیجه منفی می دهد و راست روده - نتیجه مثبت. در مرحله پیشرفته (نفروتیک) در مورد اول، نتایج در نیمی از بیماران مثبت است، و در دوم - حتی بیشتر.
در نهایت، در نارسایی مزمن کلیه، داده های بیوپسی از بافت لثه در بیش از نیمی از موارد و از مخاط رکتوم در تقریباً همه موارد مثبت است. بنابراین، بیوپسی از مخاط لثه باید برای فرآیند پیشرفته، و بیوپسی رکتوم - در هر مرحله از آمیلوئیدوز توصیه شود.
در صورت مشکوک بودن به آمیلوئیدوز ایدیوپاتیک (بیشتر با آسیب به قلب، اعصاب محیطی، کمتر - کلیه ها رخ می دهد)، اول از همه توصیه می شود که نمونه برداری از مخاط لثه انجام شود، و در صورت آمیلوئیدوز ثانویه (اکتسابی) و اشکال ارثی آن (با آسیب غالب به بیوپسی کلیه ها رخ می دهد) - a.
تعدادی از مطالعات دیگر کمک می کند: 1) تشخیص بیماری، که در برابر آن آمیلوئیدوز ایجاد شده است، روشن شود. 2) ارزیابی وضعیت عملکرد کلیه ها (آزمون Reberg، تست Zimnitsky، سطح کراتینین خون).
جریان. تصویر بالینی آمیلوئیدوز کلیه دارای ویژگی هایی است که آن را از آسیب کلیوی با منشأ متفاوت متمایز می کند: 1) سندرم نفروتیک به تدریج و اغلب پس از یک مرحله طولانی پروتئینوری ایجاد می شود، با یک دوره مداوم مشخص می شود، ادم اغلب به دیورتیک های مختلف مقاوم است. با CGN، سندرم نفروتیک، به عنوان یک قاعده، در حال حاضر در شروع بیماری رخ می دهد و اغلب در آینده عود می کند. 2) فشار خون شریانی به ندرت حتی در مرحله نارسایی مزمن کلیه مشاهده می شود. 3) در آمیلوئیدوز اولیه، نارسایی مزمن کلیه، برخلاف آمیلوئیدوز ثانویه یا CGN (به دلیل شدت کمتر آسیب گلومرولی در مقایسه با اشکال ثانویه آمیلوئیدوز) خوش خیم تر است. 4) سیر آمیلوئیدوز ثانویه تا حد زیادی به بیماری زمینه ای بستگی دارد، با تشدید مکرر که پیشرفت قابل توجه آمیلوئیدوز امکان پذیر است.
عوارض. با آمیلوئیدوز در 2-5٪ موارد ایجاد می شود:
1) ترومبوز ورید کلیه (با آمیلوئیدوز ثانویه) که آشکار می شود
هماچوری و درد در ناحیه کمر، افزایش پروتئین
ریا و کاهش دیورز؛
2) عفونت میان جریانی؛
3) پریتونیت فیبرینی-چرکی که ظاهر آن همراه است
افزایش شدید آسیت وجود دارد.
تشخیص.تظاهرات بالینی آمیلوئیدوز غیر اختصاصی است. هر یک از علائم (ادم، پروتئینوری، فشار خون شریانی) می تواند در بیماری های کلیوی مختلف رخ دهد. تنها روش برای تشخیص مطمئن آمیلوئیدوز بیوپسی از یک عضو (کلیه، کبد، مخاط رکتوم یا لثه) است، اما همیشه امکان پذیر نیست. بنابراین، در بیشتر موارد لازم است بر روی تظاهرات بالینی فرآیند پاتولوژیک تمرکز شود.
وجود یک بیماری که در آن یک ثانویه
آمیلوئیدوز (علائم بالینی یا آنامنسیک).
شروع و پیشرفت پروتئینوری یا شروع
سندرم نفروتیک
هیچ بیماری وجود ندارد که در آن آمیلوئیدوز ایجاد شود،
با این حال، پروتئینوری یا سندرم نفروتیک وجود دارد.
وجود نارسایی شدید قلبی مداوم، این سندرم نیست
کفایت جذب، پلی نوروپاتی (اگر در همان زمان آخرین باشد
توضیح این سه سندرم با دلایل دیگر دشوار است).
وجود آمیلوئیدوز را می توان با علائم آزمایشگاهی زیر مشکوک به سندرم نفروتیک (که، همانطور که می دانید، می تواند با سایر بیماری های کلیوی ایجاد شود):
الف) دیسپروتئینمی شدید + هیپوآلبومینمی + هیپر-sg- و gi-
پرگاماگلوبولینمی؛
ب) افزایش سطح ag-glycoprotein، p-lipoproteins.
ج) ظهور a- و به خصوص y-گلیکوپروتئین ها و a-lipopro- در ادرار
تیدز
در همه موارد، احتمال ایجاد آمیلوئیدوز با تشخیص کبد و طحال و همچنین تغییرات در قلب مشخصه آمیلوئیدوز افزایش می یابد (در چنین مواردی، ما در مورد آمیلوئیدوز عمومی ایدیوپاتیک صحبت می کنیم).
بنابراین، تشخیص آمیلوئیدوز کلیه را می توان با اطمینان کافی در مرحله پیشرفته (نفروتیک) یا پایانی انجام داد، در حالی که در مرحله اولیه (پروتئینوری) انجام این کار بسیار دشوارتر است. در این موارد، پروتئینوری گذرا یا دائمی باید از گلومرولونفریت (حاد، مزمن) افتراق داده شود. این باید در نظر گرفته شود:
1) پیشرفت کندتر آسیب کلیه در آمیلوئید
دوز؛
2) عدم وجود ارتباط واضح با سرماخوردگی در آمیلوئیدوز
نیامی;
3) وجود دائمی میکرو هماچوری در گلومرولونفریت (با
آمیلوئیدوز در 20 درصد موارد).
گاهی اوقات تشخیص صحیح تنها پس از یک دوره مشاهده طولانی بیمار قابل انجام است. اگر امکان انجام بیوپسی سوراخ از کلیه وجود داشته باشد، مشکل بسیار سریعتر حل می شود.
تدوین یک تشخیص بالینی دقیقآمیلوئیدوز اجزای زیر را در نظر می گیرد: 1) شکل آمیلوئیدوز. 2) مرحله آمیلوئیدوز (پروتئینوری، نفروتیک، ترمینال)؛ 3) وضعیت عملکردی کلیه ها (عدم یا وجود نارسایی کلیه، درجه شدت آن). 4) بیماری زمینه ای (با آمیلوئیدوز ثانویه)؛ 5) وضعیت سایر اندام ها (قلب، کبد، سیستم عصبی و غیره) در آمیلوئیدوز ایدیوپاتیک (اولیه).
رفتار.مشکل درمان آمیلوئیدوز هنوز حل نشده باقی مانده است، زیرا دلایل منجر به افزایش آمیلوئیدوژنز و نارسایی جذب آن مشخص نشده است. با این وجود، می توان یک سری اقدامات درمانی را انجام داد که وضعیت بیمار را بهبود می بخشد. در حال حاضر، درمان یک بیمار مبتلا به آمیلوئیدوز با در نظر گرفتن موارد زیر انجام می شود: 1) تأثیر بر بیماری زمینه ای که در برابر آن آمیلوئیدوز ایجاد شده است.
(ثانوی)؛ 2) تأثیر بر مکانیسم های پاتوژنز؛ 3) تأثیر بر سندرم های بالینی اصلی.
تأثیر بر بیماری زمینه ای که در برابر آن ایجاد می شود
آمیلوئیدوز ثانویه، با توجه به اینکه مکرر است ضروری است
استریا یا فعالیت زیاد فرآیند پاتولوژیک منجر به
پیشرفت آمیلوئیدوز
این تأثیر به شرح زیر است:
الف) در عفونت های مزمن (سل، سیفلیس)
درمان خاص طولانی مدت؛
ب) در بیماری های مزمن غیراختصاصی ریه - com
پلکس درمانی با آنتی بیوتیک، درناژ برونش و
در صورت لزوم، و مداخله جراحی (به عنوان مثال، با مزمن
آبسه ریه)؛
ج) با بیماری های سیستمیک بافت همبند، به عنوان مثال، با
آرتریت روماتوئید، درمان پیچیده نشان داده شده است، از جمله
ارزش آماده سازی های اساسی (D-penicillamine، نمک های طلا، آمینو-
داروهای نولین).
تأثیر بر مکانیسم های پاتوژنز شامل کاهش است
سنتز آمیلوئید:
الف) مصرف روزانه 80-120 گرم جگر خام به مدت 6-12 ماه
منجر به کاهش پروتئینوری، کاهش اندازه کبد می شود
نه طحال؛
ب) آماده سازی آمینوکینولین (هینگامین، یا دلاژیل، بر اساس
0.25 - 0.5 گرم در روز برای چندین ماه و حتی سالها)
yut پیشرفت فرآیند ظاهراً این ابزار نفوذ است
yut بر سنتز فیبریل های آمیلوئید. درمان فقط موثر است
در مراحل اولیه آمیلوئیدوز؛ در یک فرآیند پیشرفته
(سندرم نفروتیک مفصل، نارسایی کلیه)
نس) تجویز این داروها غیر عملی است.
ج) با ایجاد آمیلوئیدوز به دلیل بیماری دوره ای
کلشی سین توصیه می شود.
د) در آمیلوئیدوز اولیه ملفالان نیز تجویز می شود که مهار می کند
عملکرد برخی از کلون های لنفوسیت ها، به ویژه syn
تایپ ایمونوگلوبولین های زنجیره سبک درگیر در
تشکیل یک فیبریل آمیلوئید (این نیز مربوط به
آمیلوئیدوز، که در مولتیپل میلوما ایجاد می شود).
تاثیر بر سندرم های بالینی اصلی را شامل می شود
از بین بردن ادم، فشار خون بالا، و همچنین اقدامات مورد نظر
مبارزه با ایجاد نارسایی کلیه:
الف) با ایجاد سندرم نفروتیک و ادم شدید
مقدار کافی پروتئین در غذا لازم است، کاهش آن
نمک آب پز، و همچنین معرفی خون کامل یا گلبول قرمز
توده noy (به ویژه در حضور کم خونی)، استفاده دقیق
استفاده از دیورتیک ها؛
ب) فشار خون شریانی به ندرت در آمیلوئیدوز رخ می دهد،
با این حال، زمانی که به اعداد بالا می رسد، باید تعیین شود
استفاده از داروهای ضد فشار خون در انواع مختلف؛
ج) با ایجاد نارسایی کلیه، درمان طبق برنامه پذیرفته شده عمومی (محدودیت پروتئین در غذا، مصرف مایعات کافی، اصلاح متابولیسم مواد معدنی) انجام می شود. در نارسایی کلیوی ناشی از آمیلوئیدوز، می توان از همودیالیز و پیوند کلیه استفاده کرد.
پیش بینی.تعیین مدت دوره پروتئینوری دشوار است، با این حال، پس از تشخیص آن، ادم معمولا پس از 3 سال ایجاد می شود، که در مقابل آن CRF به سرعت ایجاد می شود. همه اینها پیش بینی را کاملا جدی می کند.
جلوگیری.در آمیلوئیدوز ایدیوپاتیک و ژنتیکی، اقدامات پیشگیری اولیه ناشناخته است. در آمیلوئیدوز ثانویه، پیشگیری شامل درمان بیماری هایی است که منجر به ایجاد آمیلوئیدوز می شود.
RCHD (مرکز جمهوری برای توسعه سلامت وزارت بهداشت جمهوری قزاقستان)
نسخه: پروتکل های بالینی وزارت بهداشت جمهوری قزاقستان - 2016
آمیلوئیدوز (E85)
نفرولوژی
اطلاعات کلی
توضیح کوتاه
تایید شده
کمیسیون مشترک کیفیت خدمات پزشکی
وزارت بهداشت و توسعه اجتماعی جمهوری قزاقستان
به تاریخ 13 اکتبر 2016
پروتکل شماره 13
آمیلوئیدوز- گروهی از بیماری ها که مشخصه آنها رسوب گلیکوپروتئین فیبریلار - آمیلوئید در بافت ها و اندام ها است.
همبستگی بین کدهای ICD-10 و ICD-9
| ICD-10 | ICD-9 | ||
| کد | نام | کد | نام |
| E85 | آمیلوئیدوز |
55.23 99.76 |
بیوپسی کلیه بسته [از راه پوست] [پونکشی]. همودیالیز پلاسمافرزیس درمانی جذب ایمنی خارج از بدن |
| E85.0 | آمیلوئیدوز خانوادگی ارثی بدون نوروپاتی | ||
| E85.1 | آمیلوئیدوز ارثی نوروپاتیک | ||
| E85.2 | آمیلوئیدوز ارثی، نامشخص | ||
| E85.3 | آمیلوئیدوز سیستمیک ثانویه | ||
| E85.4 | آمیلوئیدوز محدود | ||
| E85.8 | سایر اشکال آمیلوئیدوز | ||
| E85.9 | آمیلوئیدوز، نامشخص | ||
تاریخ توسعه / بازنگری پروتکل: 2016.
کاربران پروتکل:پزشکان عمومی، درمانگران، هماتولوژیست ها، نفرولوژیست ها.
مقیاس سطح شواهد:
| آ | متاآنالیز با کیفیت بالا، بررسی سیستماتیک RCTها، یا RCTهای بزرگ با احتمال بسیار کم (++) سوگیری که نتایج آنها را می توان به یک جمعیت مناسب تعمیم داد. |
| که در | بررسی سیستماتیک با کیفیت بالا (++) مطالعات همگروهی یا مورد شاهدی یا مطالعات کوهورت یا مورد شاهدی با کیفیت بالا (++) با خطر سوگیری بسیار کم یا RCTها با خطر سوگیری کم (+) که نتایج آنها را می توان به جمعیت مناسب تعمیم داد. |
| با |
کارآزمایی کوهورت یا مورد شاهدی یا کنترلشده بدون تصادفیسازی با خطر کم سوگیری (+). که نتایج آن را می توان به جمعیت مناسب یا RCT هایی با خطر سوگیری بسیار کم یا کم (++ یا +) تعمیم داد که نتایج آن را نمی توان مستقیماً به جمعیت مناسب تعمیم داد. |
| D | شرح یک سری پرونده یا مطالعه کنترل نشده یا نظر کارشناسی. |
طبقه بندی
انواع آمیلوئید و اشکال مربوط به آمیلوئیدوز:
|
پروتئین آمیلوئید |
پروتئین پیش ساز | شکل بالینی آمیلوئیدوز |
| AA | پروتئین SAA | آمیلوئیدوز ثانویه در بیماری های التهابی مزمن، از جمله بیماری های دوره ای و سندرم ماکل ولز |
| AL | λ، κ-زنجیره های سبک ایمونوگلوبولین ها | آمیلوئیدوز در دیسکرازی های پلاسما سل - ایدیوپاتیک، در مولتیپل میلوما و ماکروگلوبولینمی والدنستروم |
| ATTR | ترانس تیرتین | اشکال خانواده آمیلوئیدوز پلی نوروپاتیک، کاردیوپاتیک و سایر آمیلوئیدوز، آمیلوئیدوز سالمند سیستمیک |
| Aβ2M | β2-میکروگلوبولین | آمیلوئیدوز دیالیز |
| AGEl | ژلسولین | پلی نوروپاتی آمیلوئید خانوادگی فنلاندی |
| AApoAI | آپولیپوپروتئین A-I | پلی نوروپاتی آمیلوئید (نوع III، طبق گفته ون آلن، 1956) |
| AFib | فیبرینوژن | نفروپاتی آمیلوئید |
| Aβ | بتا پروتئین | بیماری آلزایمر، سندرم داون، خونریزی های ارثی مغزی همراه با آمیلوئیدوز، هلند |
| APrP Scr | پروتئین پریون | بیماری کروتسفلد-ژاکوب، بیماری گرستمن-استراسلر-شاینکر |
| AANF | فاکتور ناتریورتیک دهلیزی | آمیلوئیدوز دهلیزی جدا شده |
| AIAPP | آمیلین | آمیلوئیدوز جدا شده در جزایر لانگرهانس دیابتنوع دوم، انسولینوم |
| ACal | پروکلسی تونین | برای سرطان مدولاری غده تیروئید |
| ACys | سیستاتین C | خونریزی های ارثی مغزی با آمیلوئیدوز، ایسلند |
طبقه بندی بالینی آمیلوئیدوز
آمیلوئیدوز اولیه:
بدون هیچ دلیل مشخصی رخ می دهد؛
مرتبط با مولتیپل میلوما
آمیلوئیدوز ثانویه:
در عفونت های مزمن؛
در آرتریت روماتوئید و سایر بیماری های بافت همبند؛
در بیماری های انکولوژیک؛
آمیلوئیدوز خانوادگی (ارثی):
در صورت بیماری دوره ای؛
نوع پرتغالی و سایر اشکال آمیلوئیدوز خانوادگی؛
آمیلوئیدوز پیری
آمیلوئیدوز موضعی
آمیلوئیدوز ارثی:
عصبی
با آسیب به اندام تحتانی: پرتغالی، ژاپنی، سوئدی و انواع دیگر.
با ضایعات اندام فوقانی: انواع سوئیس-ایندیانا، آلمان-مریلند؛
نفروپاتی:
بیماری دوره ای
تب و درد شکم در سوئدی ها و سیسیلی ها.
ترکیبی از راش، ناشنوایی و آسیب کلیه؛
آسیب کلیه در ترکیب با فشار خون شریانی؛
کاردیومیوپاتیک:
دانمارکی - نارسایی قلبی پیشرونده؛
· مکزیکی-آمریکایی - سندرم سینوس بیمار، ایست دهلیزی.
مختلط:
فنلاندی - دیستروفی قرنیه و آسیب به اعصاب جمجمه.
سکته های مغزی
مراحل بالینی آمیلوئیدوز کلیه
| صحنه | تظاهرات بالینی |
| 1 | مرحله پیش بالینی یا نهفته (بدون علامت) - آمیلوئید در ناحیه میانی وجود دارد و ادم و کانون های اسکلروز در امتداد عروق مستقیم اهرام ایجاد می شود. مرحله 3-5 سال یا بیشتر طول می کشد. در این دوره، آمیلوئیدوز واکنشی توسط تظاهرات بالینی بیماری زمینه ای (مانند فرآیند چرکی در ریه ها، سل، آرتریت روماتوئید و غیره) غالب می شود. |
| 2 | مرحله پروتئینی (آلبومینوریک) - آمیلوئید در درجه اول در مزانژیوم، در حلقه های مویرگ ها، در اهرام و ماده قشر گلومرول ها، در عروق ظاهر می شود. اسکلروز و آتروفی نفرون ها، هیپرمی و لنفوستاز ایجاد می شود. کلیه ها بزرگ و متراکم، به رنگ خاکستری مایل به صورتی مات هستند. پروتئینوری در ابتدا به طور متوسط بیان می شود، حتی می تواند برای مدتی گذرا باشد، کاهش و افزایش یابد، اما پس از آن پایدار می شود (مرحله پروتئینوری متناوب). برخی از محققان در این مرحله دو دوره را تشخیص می دهند: پروتئینوری انتخابی و غیر انتخابی. مدت زمان مرحله از 10 تا 13 سال است. |
| 3 | مرحله نفروتیک (ادماتوز، ادماتوز-هیپوتونیک) - نفروز آمیلوئید - لیپوئید - آمیلوئید در تمام قسمتهای نفرون. اسکلروز و آمیلوئیدوز مدولا وجود دارد، اما لایه قشر بدون تغییرات اسکلروتیک مشخص است. مدت زمان مرحله تا 6 سال است. هم در مرحله پروتئینوریک و هم در مرحله نفروتیک، کلیه ها بزرگ و متراکم هستند (کلیه سباسه بزرگ). از نظر بالینی، این مرحله با سندرم نفروتیک کلاسیک با تمام علائم خود ظاهر می شود: با ایجاد پروتئینوری عظیم (با از دست دادن پروتئین در ادرار بیش از 3-5 گرم در روز)، هیپوپروتئینمی با هیپوآلبومینمی، هیپرکلسترولمی، لیپیدوری با ادم تا درجه آنسارکا. گچ های هیالین در رسوبات ادراری یافت می شود و با افزایش پروتئینوری، قالب های دانه ای یافت می شود. میکرو و ماکرو هماچوری احتمالی، لکوسیتوری بدون علائم پیلونفریت. |
| 4 | مرحله اورمیک (ترمینال، آزوتمیک) - کلیه چروکیده آمیلوئید - کاهش اندازه، متراکم، کلیه اسکار. نارسایی مزمن کلیه تفاوت کمی با سایر بیماری های کلیوی دارد. اعتقاد بر این است که بر خلاف گلومرولونفریت، که در آن شروع CRF با پلی اوری می تواند منجر به حداقل همگرایی نسبی ادم شود، در آمیلوئیدوز، آزوتمی در پس زمینه فشار خون پایین و سندرم نفروتیک ایجاد می شود. |
تشخیص (کلینیک سرپایی)
تشخیص در سطح سرپایی
معیارهای تشخیصی
شکایات:
ضعف، افزایش خستگی؛
· سردرد؛
تورم در پاها، بازوها و صورت؛
فشار خون بالا؛
حالت تهوع، اسهال (اسهال)؛
درد در ناحیه قلب؛
درد عضلانی
تاریخچه:
· کاهش وزن؛
وجود گاموپاتی مونوکلونال با منشا ناشناخته؛
بیماری های التهابی مزمن (چرکی)؛
عفونت های مزمن؛
وراثت
معاینهی جسمی
بازرسی عمومی:
پورپورای اطراف چشم (در 15٪ موارد مشاهده شد).
macroglossia مشخصه آمیلوئیدوز اولیه (AL) است.
تنگی نفس در طی فعالیت بدنی (در حدود 40٪ از بیماران مشاهده شد).
علامت شانه (انفیلتراسیون اطراف مفصلی آمیلوئید منجر به هیپرتروفی کاذب و افزایش حجم عضلات کمربند شانه و ران می شود).
سمع:
وجود احتمالی آریتمی قلبی
لمس:
تورم اندام تحتانی به دلیل هیپوآلبومینمی و سندرم نفروتیک و همچنین به دلیل رکود در گردش خون سیستمیک به دلیل کاردیومیوپاتی محدود کننده (در 50٪ موارد مشاهده شد).
بزرگ شدن کبد و طحال؛
پارستزی (در حدود 15٪ بیماران مشاهده شد)؛
درد اسپاستیک در دستگاه گوارش؛
ممکن است در غدد بزاقی زیر فکی افزایش یابد.
تحقیقات آزمایشگاهی:
شمارش کامل خون - کم خونی، لکوسیتوز، افزایش ESR.
تجزیه و تحلیل کلی ادرار - پروتئینوری، میکرو هماچوری، لکوسیتوری آسپتیک.
آزمایش خون بیوشیمیایی (پروتئین کل، آلبومین، سدیم، کلسیم، کلسترول، قند در سرم خون) - هیپوپروتئینمی (به دلیل هیپوآلبومینمی)، هیپرگلوبولینمی، هیپوناترمی، هیپوپرروترومبینمی، هیپوکلسمی، هیپرکلسترولمی.
سونوگرافی اندام ها حفره شکمیو کلیه ها - کلیه های فشرده بزرگ شده (کلیه های چرب بزرگ) مشاهده می شوند.

تشخیص (بیمارستانی)
تشخیص در سطح ثابت
معیارهای تشخیصی در سطح بیمارستان
شکایات و شرح حال:سطح سرپایی را ببینید
معاینهی جسمی:سطح سرپایی را ببینید
تحقیقات آزمایشگاهی:
| تست تشخیصی | نتیجه |
|
ایمونوفیکساسیون سرم این آزمایش در 60 درصد بیماران آمیلوئیدوز دارای ایمونوگلوبولین با زنجیره سبک (AL) مثبت است (6). |
|
|
ایمونوفیکساسیون ادرار این آزمایش در 80 درصد بیماران مبتلا به آمیلوئیدوز AL مثبت است (6). تشخیص پروتئین زنجیره سبک در ادرار حاکی از وجود مولتیپل میلوما و آمیلوئیدوز است. |
وجود یک پروتئین مونوکلونال |
|
آزمایش ایمونوگلوبولین بدون زنجیره سبک سرم این تست نسبتا جدید با بسیار حساسیت بالا(>95%) برای تشخیص آمیلوئیدوز AL (10). آنتی سرم های موجود تجاری علیه زنجیره های سبک ایمونوگلوبولین، AA و ترانس تیرتین معمولاً مورد استفاده قرار می گیرند اما ممکن است ویژگی و حساسیت کافی نداشته باشند. در بسیاری از موارد، طیفسنجی جرمی و میکروسکوپ ایمونو الکترونی برای تعیین نوع آمیلوئید زیرین مورد نیاز است. |
نسبت کاپالامبدا غیر طبیعی |
|
بیوپسی مغز استخوان
بیوپسی مغز استخوان در تمام بیماران مشکوک به آمیلوئیدوز با زنجیره سبک انجام می شود و می باشد منبع عالیبافتی برای تشخیص هر بیمار مشکوک به آمیلوئیدوز. |
وجود سلول های پلاسما کلونال |
تحقیق ابزاری:
سونوگرافی اندام های شکمی و کلیه ها - کلیه های فشرده بزرگ شده (کلیه های چرب بزرگ) مشاهده می شوند.
الگوریتم تشخیصی آمیلوئیدوز کلیه
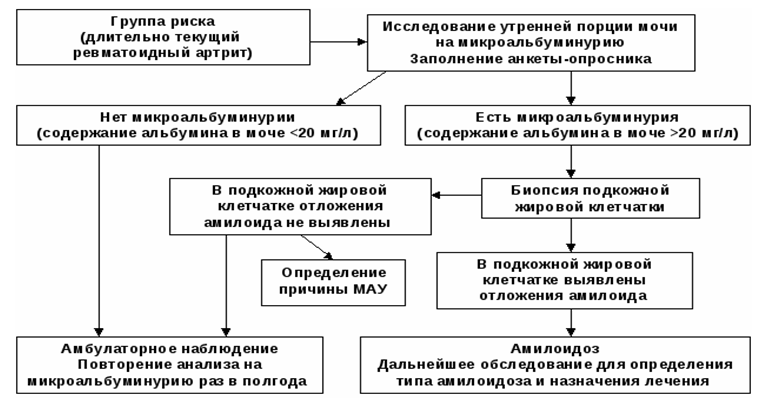
لیست اصلی اقدامات تشخیصی:
· تجزیه و تحلیل خون عمومی.
تجزیه و تحلیل کلی ادرار
تجزیه و تحلیل بیوشیمیایی خون (پروتئین کل، آلبومین، سدیم، کلسیم، کلسترول، قند در سرم خون).
ایمونوفیکساسیون سرم؛
ایمونوفیکساسیون ادرار؛
بررسی ایمونوگلوبولین های زنجیره سبک آزاد در سرم.
بیوپسی مغز استخوان
سونوگرافی از اندام های شکم و کلیه ها.
فهرست اقدامات تشخیصی اضافی
تحقیقات آزمایشگاهی:
| تست تشخیصی | نتیجه |
|
بیوپسی بافت: برای تشخیص آمیلوئیدوز، لازم است که رسوبات در بافت های موجود در ماده بیوپسی به رنگ قرمز کنگو رنگ مثبت داشته باشد (11). هنگامی که مواد کنگو تحت نور پلاریزه به رنگ قرمز رنگ می شوند، انکسار دوگانه سبز روشن دیده می شود. مواد بیوپسی را می توان از مخاط لب، پوست، لثه، چربی زیر جلدی، مغز استخوان، اعصاب، راست روده، کلیه ها، کبد یا قلب به دست آورد. رسوبات همیشه خارج سلولی قرار دارند و بی شکل هستند. |
مثبت - انکسار دوگانه سبز هنگام رنگ آمیزی با قرمز کنگو |
|
مطالعات ایمونوهیستولوژیک رسوبات آمیلوئیدی: آنها به شما امکان می دهند اشکال مختلف آمیلوئیدوز سیستمیک را تشخیص دهید. |
آنتی سرم علیه ایمونوگلوبولین های زنجیره سبک، AA و ترانس تیرتین |
|
طیف سنجی جرمی: تجزیه و تحلیل ترکیب پروتئین آمیلوئید را ارائه می دهد. در حال حاضر استاندارد طلایی برای تشخیص نوع آمیلوئیدوز است. |
نوع پروتئین را تایید می کند |
|
میکروسکوپ ایمونو الکترونی: تمام اشکال آمیلوئید میکروسکوپ الکترونیفیبری، سفت و بدون انشعاب. |
آمیلوئیدها ظاهری فیبریل دارند و سفت و بدون انشعاب هستند. |
|
آزمایش ژنتیک: آزمایش ژنتیکی برای رد آمیلوئیدوز ارثی در حضور نتایج مشکوک تست تشخیص پروتئین زنجیره سبک مونوکلونال ایمونوگلوبولین/آمیلوئید اجباری است. ژن ها را می توان با توالی یابی مستقیم بررسی کرد و شامل ژن های زیر است: TTR، فیبرینوژن، آپولیپوپروتئین A1، لیزوزیم، MEFV (تب مدیترانه ای)، و گیرنده فاکتور نکروز تومور 1 (TNFR1 یا TNFRSF1A). MEFV و TNFRSF1A سندرم های ارثی هستند تب دوره ای(به عنوان مثال، علل بالقوه آمیلوئیدوز AA)، و به خودی خود آمیلوئیدوز ارثی نیستند. |
مثبت |
|
اسکن سینتی گرافی آمیلوئید P (SAP): در سال های اخیر، سینتی گرافی با ید نشاندار شده با مولفه P (SAP) در عمل بالینی برای ارزیابی توزیع آمیلوئید در بدن مورد استفاده قرار گرفته است. |
جذب در محل های رسوب آمیلوئید |
|
تحلیل عمومیخون: کم خونی عمدتاً در بیماران مبتلا به نارسایی کلیوی یا خونریزی از دستگاه گوارش رخ می دهد. ترومبوسیتمی با درگیری کبد و هیپرسپلنیسم همراه است. |
معمولا نرمال |
|
آزمایش خون بیوشیمیایی (آزمایش کبد و کلیه), شاخص های وضعیت متابولیک): آمیلوئیدوز کبد با افزایش سطح مشخص می شود فسفاتاز قلیایی. اکثر بیماران در مراحل اولیه آمیلوئیدوز کلیه، کلیرانس کراتینین را حفظ می کنند، اما ممکن است درجات قابل توجهی از هیپوآلبومینمی به دلیل از دست دادن پروتئین در ادرار (سندرم نفروتیک) وجود داشته باشد. |
آلبومین کم؛ افزایش آلکالین فسفاتاز |
|
پروتئینوری روزانه (جمع آوری ادرار در 24 ساعت): دفع آلبومین بیش از 1 گرم در روز در بیماران مبتلا به آمیلوئیدوز نشان دهنده آسیب کلیه (آمیلوئیدوز کلیه) است. در سطح پروتئینوری بیش از 3 گرم در روز، سندرم نفروتیک ایجاد می شود. |
افزایش پروتئین در ادرار |
|
سطح سرمی تروپونین: یک آزمایش حساس برای تعیین آسیب میوکارد. بیمارانی که سطح تروپونین قابل تشخیص دارند، پیش آگهی بدتری نسبت به بیماران بدون آن دارند (12). |
مرتفع |
|
پپتید ناتریورتیک نوع B: مطالعه تشخیصی حساس برای وجود اتساع میوکارد و CHF. نشان داده شده است که مهم است ارزش پیش بینیدر ایجاد آمیلوئیدوز قلب (13). در سطح پپتید ناتریورتیک نوع B > 300 نانوگرم در لیتر (> 300 pg/mL) نشان دهنده درگیری آمیلوئید میوکارد است (10). بیماران مبتلا به<170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с >170 ng/l (> 170 pg/ml). |
مرتفع |
|
میکروگلوبولین های بتا 2: این یک پیش بینی کننده بقا در بیماران مبتلا به آمیلوئیدوز است. در سطح بتا-2 میکروگلوبولین > 2.7 میلی گرم در لیتر، پیش آگهی نامطلوب است (14). |
مرتفع |
تحقیق ابزاری:
|
نوار قلب: باید در تمام بیماران به عنوان بخشی از ارزیابی درگیری قلبی انجام شود. |
اختلال هدایت قلب |
|
اکوکاردیوگرافی (EchoCG): علائم بالینی نارسایی قلبی در بیماران مبتلا به آمیلوئیدوز قلبی از 22% تا 34% مشاهده می شود (7). اکوکاردیوگرافی شیوع بالایی از رسوب آمیلوئید را در بیماران با حداقل علائم نشان می دهد (حدود 50٪ موارد AL درگیری قلبی دارند). که در آخرین مرحلهکاهش در کسر جهشی وجود دارد. |
اختلال عملکرد دیاستولیک، ضخیم شدن سپتوم بین بطنی، کاهش کسر جهشی |
|
اکو داپلر همراه با کشش: نشانگر درجه نفوذ آمیلوئید در میوکارد. هنگامی که فشار خون شریانی یا بیماری دریچه ای قلب وجود ندارد، حساسیت بالایی در تشخیص ناهنجاری ها دارد. کشش میوکارد به عنوان درصد تغییر طول فیبر میوکارد در واحد طول تعریف می شود و میزان آن به مدت کشش بستگی دارد (15-16). |
کاهش انقباض طولی و کشش میوکارد؛ محدودیت پر شدن بطن |
|
ام آر آی قلب: ریلکسومتری رزونانس مغناطیسی قابلیت اطمینان تشخیص MRI را بهبود می بخشد و به تشخیص آمیلوئیدوز قلبی از کاردیومیوپاتی هیپرتروفیک کمک می کند. |
به طور قابل توجهی زمان آرامش T1 و T2 را در مقایسه با گروه کنترل افزایش داد |
تشخیص های افتراقی
تشخیص های افتراقیآمیلوئیدوز کلیه
| حالت | علائم/نشانه های متمایز | تست های متفاوت |
| کاردیومیوپاتی هیپرتروفیک | از نظر بالینی تشخیص HCM از آمیلوئیدوز قلبی دشوار است. |
اکوکاردیوگرافی است معیار تشخیصیبرای GCM، که در آن هیپرتروفی نامتقارن سپتوم بین بطنی تشخیص داده می شود. اکو داپلر همراه با کشش، که برای حذف علائم آمیلوئیدوز استفاده میشود، نشاندهنده تغییرات پرکننده محدود کننده معمولی شناساییشده در آمیلوئیدوز نیست. MRI می تواند 2 سندرم را تشخیص دهد |
| گلومرولوپاتی غشایی | تظاهرات بالینی یکسان در بیماران مبتلا به سندرم نفروتیک. | بیوپسی کلیه با قرمز کنگو رنگ نمی شود. |
| گاموپاتی مونوکلونال با منشا ناشناخته (MGNG) نوروپاتی مرتبط | بیماران درجه قابل توجهی از پروتئینوری، هپاتومگالی یا کاردیومیوپاتی ندارند. | بیوپسی عصب سورال با قرمز کنگو رنگ نمی شود. |
| مولتیپل میلوما | درد استخوان، علائم کم خونی و نارسایی کلیه. |
معمولی اشعه ایکسنشان دادن ضایعات استخوان لیتیک، شکستگی های فشاریپوکی استخوان منتشر; Hb پایین؛ نارسایی کلیه. |
| سندرم نفروتیک | پروتئینوری روزانه بیش از 3.5 گرم در روز، ادم، هیپوآلبومینمی، دیس لیپیدمی | KP "سندرم نفروتیک" را ببینید |
گردشگری پزشکی
دریافت درمان در کره، اسرائیل، آلمان، ایالات متحده آمریکا
گردشگری پزشکی
در مورد گردشگری پزشکی مشاوره بگیرید
رفتار
توجه!
- با خوددرمانی می توانید آسیب های جبران ناپذیری به سلامتی خود وارد کنید.
- اطلاعات ارسال شده در وب سایت MedElement نمی تواند و نباید جایگزین شود مشاوره حضوریدکتر حتما تماس بگیرید موسسات پزشکیاگر بیماری یا علائمی دارید که شما را آزار می دهد.
- انتخاب داروها و میزان مصرف آنها باید با متخصص مشورت شود. فقط پزشک می تواند داروی مناسب و دوز آن را با در نظر گرفتن بیماری و وضعیت بدن بیمار تجویز کند.
- وب سایت MedElement فقط یک منبع اطلاعاتی و مرجع است. اطلاعات درج شده در این سایت نباید برای تغییر خودسرانه نسخه پزشک استفاده شود.
- سردبیران MedElement هیچ مسئولیتی در قبال آسیب به سلامتی یا آسیب مادی ناشی از استفاده از این سایت ندارند.
- عمومی، بیماری سیستمیکارگانیسمی که در آن یک گلیکوپروتئین خاص (آمیلوئید) در اندامها و بافتها با اختلال در عملکرد دومی رسوب میکند. با آمیلوئیدوز، کلیه ها (سندرم نفروتیک، سندرم ادماتوز)، قلب (نارسایی قلبی، آریتمی)، دستگاه گوارش، سیستم اسکلتی عضلانی و پوست می توانند تحت تأثیر قرار گیرند. توسعه احتمالی پلی سروزیت، سندرم هموراژیک، اختلالات روانی. تشخیص مطمئن آمیلوئیدوز با تشخیص آمیلوئید در نمونه های بیوپسی بافت های آسیب دیده تسهیل می شود. برای درمان آمیلوئیدوز، درمان سرکوب کننده سیستم ایمنی و علامتی انجام می شود. با توجه به نشانه ها - دیالیز صفاقی، پیوند کلیه و کبد.
ICD-10
E85

اطلاعات کلی
آمیلوئیدوز یک بیماری از گروه دیسپروتئینوزهای سیستمیک است که با تشکیل و تجمع در بافت های یک ترکیب پروتئین-پلی ساکارید پیچیده - آمیلوئید رخ می دهد. شیوع آمیلوئیدوز در جهان تا حد زیادی از نظر جغرافیایی تعیین می شود: برای مثال، بیماری دوره ای در کشورهای حوزه مدیترانه شایع تر است. پلی نوروپاتی آمیلوئید - در ژاپن، ایتالیا، سوئد، پرتغال و غیره. فرکانس متوسطآمیلوئیدوز در جمعیت 1 مورد در 50 هزار نفر است. این بیماری معمولا در افراد مسن تر از 50-60 سال ایجاد می شود. با توجه به این واقعیت که تقریباً تمام سیستم های اندام در آمیلوئیدوز تحت تأثیر قرار می گیرند، این بیماری توسط رشته های مختلف پزشکی مورد مطالعه قرار می گیرد: روماتولوژی، اورولوژی، قلب و عروق، گوارش، نورولوژی و غیره.
علل آمیلوئیدوز
علت آمیلوئیدوز اولیه به طور کامل شناخته نشده است. در عین حال، مشخص شده است که آمیلوئیدوز ثانویه معمولاً با بیماری های عفونی مزمن (سل، سیفلیس، اکتینومیکوز) و بیماری های التهابی چرکی (استئومیلیت، برونشکتازی، اندوکاردیت باکتریایی و غیره) همراه است، کمتر - فرآیندهای تومور(لنفوگرانولوماتوز، لوسمی، سرطان اندام های احشایی). آمیلوئیدوز واکنشی می تواند در بیماران مبتلا به آترواسکلروز، پسوریازیس، روماتولوژی (آرتریت روماتوئید، اسپوندیلیت آنکیلوزان) ایجاد شود. التهاب مزمن(کولیت اولسراتیو، بیماری کرون)، ضایعات چند سیستمی (بیماری ویپل، سارکوئیدوز). از جمله عوامل موثر در ایجاد آمیلوئیدوز، هیپرگلوبولینمی، اختلال در عملکرد ایمنی سلولی، استعداد ژنتیکی و غیره از اهمیت بالایی برخوردار است.
پاتوژنز
در میان بسیاری از نسخه های آمیلوئیدوژنز بیشترین تعدادطرفداران نظریه دیسپروتئینوزیس، پیدایش سلولی موضعی، نظریه های ایمونولوژیک و جهش دارند. تئوری پیدایش سلولی محلی تنها فرآیندهایی را در نظر می گیرد که در سطح سلولی (تشکیل پیش سازهای آمیلوئید فیبریلار توسط سیستم ماکروفاژها) اتفاق می افتد، در حالی که تشکیل و تجمع آمیلوئید در خارج از سلول اتفاق می افتد. بنابراین، نظریه پیدایش سلولی محلی را نمی توان جامع در نظر گرفت.
بر اساس تئوری دیسپروتئینوز، آمیلوئید محصول متابولیسم غیر طبیعی پروتئین است. پیوندهای اصلی در پاتوژنز آمیلوئیدوز - دیسپروتئینمی و هیپرفیبرینوژنمی به تجمع پروتئین درشت و فراپروتئین در پلاسما کمک می کند. تئوری ایمونولوژیک منشا آمیلوئیدوز تشکیل آمیلوئید را با واکنش آنتی ژن-آنتی بادی مرتبط می کند که در آن پروتئین های خارجی یا محصولات پوسیده بافت های خود به عنوان آنتی ژن عمل می کنند. در این مورد، رسوب آمیلوئید عمدتا در مکان های تشکیل آنتی بادی ها و آنتی ژن های اضافی رخ می دهد. جهانی ترین نظریه جهش آمیلوئیدوز است که طیف عظیمی از عوامل جهش زا را در نظر می گیرد که می توانند باعث سنتز غیر طبیعی پروتئین شوند.
آمیلوئید یک گلیکوپروتئین پیچیده متشکل از پروتئین های فیبریلار و کروی است که با پلی ساکاریدها مرتبط است. رسوبات آمیلوئید در انتیما و آدونتیتای رگ های خونی، استرومای اندام های پارانشیمی، ساختارهای غده ای و ... تجمع می یابند. با رسوبات جزئی آمیلوئید، تغییرات فقط در سطح میکروسکوپی تشخیص داده می شود و منجر به اختلالات عملکردی نمی شود. تجمع مشخص آمیلوئید با تغییرات ماکروسکوپی در اندام آسیب دیده (افزایش حجم، ظاهر چرب یا مومی شکل) همراه است. در نتیجه آمیلوئیدوز، اسکلروز استرومایی و آتروفی پارانشیم اندام ها، کمبود عملکردی قابل توجه بالینی آنها ایجاد می شود.
طبقه بندی
بر اساس علل، آمیلوئیدوز اولیه (ایدیوپاتیک)، ثانویه (واکنشی، اکتسابی)، ارثی (خانوادگی، ژنتیکی) و آمیلوئیدوز پیری تشخیص داده می شود. انواع مختلفی از آمیلوئیدوز ارثی وجود دارد: تب مدیترانهای یا بیماری دورهای (حملات گرم، درد شکم، یبوست، اسهال، پلوریت، آرتریت، بثورات پوستی)، آمیلوئیدوز نوروپاتیک پرتغالی (پلی نوروپاتی محیطی، ناتوانی جنسی، بیماریهای عصبی واریفیای قلبی، اختلالات هدایت عصبی واریسی کاردیو کاریک فنلاندی)، یلوئیدوز) و pl. دیگران
بسته به آسیب غالب به اندام ها و سیستم ها، نفروپاتیک (آمیلوئیدوز کلیه ها)، کاردیوپاتیک (آمیلوئیدوز قلب)، نوروپاتیک (آمیلوئیدوز سیستم عصبی)، هپاتوپاتیک (آمیلوئیدوز کبد)، اپی نفروپاتیک (آمیلوئیدوز غدد آدرنال و نوع پوستی آمیلوئیدوز APUD)، APUD هستند. علاوه بر این، در عمل بین المللی مرسوم است که بین آمیلوئیدوز موضعی و عمومی (سیستمیک) تمایز قائل شود. به شکل های محلی، به عنوان یک قاعده، در افراد ایجاد می شود کهنسالشامل آمیلوئیدوز در بیماری آلزایمر، دیابت نوع 2، تومورهای غدد درون ریز، تومورهای پوستی، مثانهو غیره بسته به ترکیب بیوشیمیاییفیبرهای آمیلوئید در بین اشکال سیستمیک آمیلوئیدوز، انواع زیر متمایز می شوند:
- AL- به عنوان بخشی از فیبریل ها، زنجیره های سبک Ig (با بیماری والدنستروم، مولتیپل میلوم، لنفوم بدخیم).
- AA- به عنوان بخشی از فیبریل ها، α-گلوبولین سرم فاز حاد، شبیه به پروتئین واکنشی C (برای بیماری های تومور و روماتیسمی، بیماری های دوره ای و غیره).
- Aβ2M- به عنوان بخشی از فیبرهای β2-میکروگلوبولین (در نارسایی مزمن کلیه در بیماران تحت همودیالیز).
- ATTR- در ترکیب فیبریل ها، پروتئین انتقال دهنده ترانس تیرتین (در اشکال ارثی و پیری خانوادگی آمیلوئیدوز).
علائم آمیلوئیدوز
تظاهرات بالینی آمیلوئیدوز متنوع است و به شدت و محل رسوبات آمیلوئید، ترکیب بیوشیمیایی آمیلوئید، "تجربه" بیماری و درجه اختلال عملکرد اندام بستگی دارد. در مرحله نهفته آمیلوئیدوز، زمانی که رسوبات آمیلوئید را فقط می توان از طریق میکروسکوپی تشخیص داد، هیچ علامتی وجود ندارد. با توسعه و پیشرفت نارسایی عملکردی یک یا آن اندام، علائم بالینی بیماری افزایش می یابد.
با آمیلوئیدوز کلیه، مرحله فعلی طولانی مدت پروتئینوری متوسط با ایجاد سندرم نفروتیک جایگزین می شود. انتقال به مرحله پیشرفته ممکن است با یک عفونت داخلی، واکسیناسیون، هیپوترمی، تشدید بیماری زمینه ای همراه باشد. ادم به تدریج افزایش می یابد (اول در پاها و سپس در کل بدن)، فشار خون شریانی نفروژنیک و نارسایی کلیه ایجاد می شود. ممکن است ترومبوز سیاهرگ کلیوی رخ دهد. از دست دادن انبوه پروتئین با هیپوپروتئینمی، هیپرفیبرینوژنمی، هیپرلیپیدمی و آزوتمی همراه است. در ادرار، میکرو، گاهی اوقات ماکرو هماچوری، لکوسیتوری یافت می شود. به طور کلی، در طول آمیلوئیدوز کلیه، مرحله اولیه غیر ادماتوز، مرحله ادماتوز و مرحله اورمیک (کشکتیک) تشخیص داده می شود.
آمیلوئیدوز قلب با توجه به نوع کاردیومیوپاتی محدود کننده با علائم بالینی معمولی - کاردیومگالی، آریتمی، نارسایی پیشرونده قلب پیش می رود. بیماران از تنگی نفس، تورم، ضعفی که با فعالیت بدنی جزئی رخ می دهد شکایت دارند. در موارد کمتر، با آمیلوئیدوز قلب، پلی سروزیت (آسیت، پلوریت اگزوداتیو و پریکاردیت) ایجاد می شود.
شکست دستگاه گوارش در آمیلوئیدوز با نفوذ آمیلوئید زبان (ماکروگلاسیا)، مری (سفتی و اختلال در پریستالسیس)، معده (سوزش سر دل، تهوع)، روده ها (یبوست، اسهال، سندرم انسداد سوء جذب در تست) مشخص می شود. خونریزی دستگاه گوارش ممکن است در سطوح مختلف رخ دهد. با انفیلتراسیون آمیلوئید کبد، هپاتومگالی، کلستاز و فشار خون پورتال ایجاد می شود. درگیری پانکراس در آمیلوئیدوز معمولا به عنوان پانکراتیت مزمن ظاهر می شود.
آمیلوئیدوز پوست با ظهور پلاک های مومی متعدد (پاپول ها، ندول ها) در صورت، گردن، چین های طبیعی پوست رخ می دهد. از نظر خارجی، ضایعات پوستی ممکن است شبیه اسکلرودرمی، نورودرماتیت یا لیکن پلان باشند. برای ضایعات آمیلوئید سیستم اسکلتی عضلانی، ایجاد پلی آرتریت متقارن، کارپال سندرم تونل, پری آرتریت استخوان بازو , میوپاتی . فرم های جداآمیلوئیدوز، که با درگیری سیستم عصبی رخ می دهد، ممکن است با پلی نوروپاتی، فلج اندام تحتانی، سردرد، سرگیجه، افت فشار خون ارتواستاتیک، تعریق، زوال عقل و غیره همراه باشد.
تشخیص
پزشکان مختلف ممکن است با تظاهرات بالینی آمیلوئیدوز مواجه شوند: روماتولوژیست ها، اورولوژیست ها، قلب و عروق، متخصصین گوارش، متخصصان مغز و اعصاب، متخصصین پوست، درمانگران و غیره. تنظیم صحیحتشخیص دارای یک ارزیابی جامع از علائم بالینی و آنامنسیک، یک آزمایشگاه جامع و معاینه ابزاری است.
آمیلوئیدوز یک بیماری سیستمیک است که در آن متابولیسم پروتئین، سیستم ایمنی از کار می ایستد. در این راستا، آمیلوئید تشکیل می شود - یک مجموعه پروتئین-ساکارید که در تمام بافت های اندام های انسان رسوب می کند.
با گذشت زمان، آمیلوئید بیشتر و بیشتر به اندام ها حمله می کند و سلول های طبیعی را از بین می برد. در نتیجه، اندام عملکرد خود را از دست می دهد، وجود دارد تغییرات غیر قابل برگشت. اگر بیماری درمان نشود برای مدت طولانی، عملکرد چندین اندام نقض می شود که منجر به مرگ می شود.
طبق تحقیقات WHO، آمیلوئیدوز در حدود 1٪ از ساکنان جهان تشخیص داده می شود. شایع ترین آمیلوئیدوز ثانویه است. آمیلوئیدوز ژنتیکی اغلب در افراد متعلق به ملیت یهودی، ارمنی و همچنین در ساکنان کشورهای حوزه مدیترانه تشخیص داده می شود.
میزان بروز در مردان دو برابر زنان است. در بین انواع آمیلوئیدوز، آمیلوئیدوز نفروپاتیک (آسیب کلیه) و عمومی (آسیب به تمام بافت ها و اندام ها) تشخیص داده می شود.
انواع آمیلوئیدوز، علل ایجاد
بسته به علت آمیلوئیدوز، انواعی از بیماری وجود دارد که می تواند به طور مستقل یا به دلیل آسیب شناسی در سایر سیستم ها و اندام ها ایجاد شود. ملاقات انواع زیرآمیلوئیدوز: پیری، با تومور، آمیلوئیدوز اولیه یا ایدیوپاتیک، ارثی، ثانویه یا واکنشی، و همچنین در بیماران تحت همودیالیز. بسته به گونه، توسعه آمیلوئیدوز متفاوت است، علائم و پیش آگهی متفاوت است. انواع و مراحل آمیلوئیدوز در زیر به تفصیل مورد بحث قرار خواهد گرفت.
اولیه (ایدیوپاتیک)
آمیلوئیدوز اولیه در بیشتر موارد بدون دلیل شروع می شود. با این شکل از بیماری، آمیلوئید در بافت ها و اندام ها رسوب می کند و جهش سلول های سیستم ایمنی مشاهده می شود. AL-آمیلوئید تشکیل شده در این فرآیند در عضلات، پوست، سیستم قلبی عروقی و اعصاب تجمع می یابد. همچنین، AL-آمیلوئید در پس زمینه میلوم تومور، زمانی که سلول های پلاسما شروع به ترشح گلوبولین ها در حجم زیادی می کنند، تشکیل می شود. پس از اتصال به نوکلئوپروتئین های پلاسما، گلوبولین های غیر طبیعی به آمیلوئید تبدیل می شوند.
ثانویه (واکنشی)
آمیلوئیدوز ثانویه در پس زمینه فرآیندهای التهابی پیشرونده در طول زمان ایجاد می شود. در این حالت، AA-آمیلوئید به عنوان عارضه سایر بیماری ها تشکیل می شود. علل آمیلوئیدوز ثانویه عبارتند از:
- عفونت های مزمن - جذام، مالاریا، سل، سیفلیس، پیلونفریت، برونشکتازی.
- بیماری های مزمن چرکی - چرکی زخم ها برای مدت طولانی، استئومیلیت.
- تومورها - لوسمی، لنفوگرانولوماتوز و غیره.
- وجود کولیت اولسراتیو غیر اختصاصی (التهاب روده بزرگ).
- بیماری روماتولوژیک - اسپوندیلیت آنکیلوزان، آرتریت روماتوئید و غیره.
آمیلوئیدوز ثانویه می تواند بر هر اندام، بافتی در بدن تأثیر بگذارد. تظاهرات تصویر بیماری بلافاصله قابل توجه نیست. سالها پس از شروع بیماری زمینهای، میتوان متوجه نقض عملکرد ارگانی شد که بیشتر از همه در آن آمیلوئید رسوب کرده است. بیشتر اوقات، کبد، کلیه ها، طحال و غدد لنفاوی تحت تأثیر این اختلال قرار می گیرند. با گذشت زمان، سایر اندام ها نیز تحت تأثیر قرار می گیرند که منجر به نارسایی چند عضوی و مرگ می شود.
آمیلوئیدوز ارثی
شکل ارثی آمیلوئیدوز به دلیل وجود ژن های جهش یافته در سلول های سیستم ایمنی است. این جهش های ژنتیکی از طریق نسل ها منتقل می شوند و در نتیجه آمیلوئیدوبلاست ها تشکیل می شوند. شکل ارثی افراد در یک منطقه خاص یا متعلق به یک منطقه خاص را تحت تاثیر قرار می دهد گروه قومی. آمیلوئیدوز ارثی به انواع زیر تقسیم می شود:
- کاردیوپاتیک. بیشتر در ساکنان دانمارک تشخیص داده می شود. تصویر بالینی بیماری شبیه آمیلوئیدوز اولیه از نوع عمومی است.
- عصبی با شکست مشخص می شود بافت عصبی. بسته به محل ضایعه، آمیلوئیدوز پرتغالی (اعصاب پاها)، آمریکایی (اعصاب دست ها)، فنلاندی (سیستم عصبی، قرنیه چشم، کلیه ها) وجود دارد.
- نفروپاتی خانوادگی نام دیگر آمیلوئیدوز انگلیسی (بیماری ماکل و ولز) است. تصویر بالینی کهیر، حملات تب، کاهش شنوایی است.
- دوره ای (تب مدیترانه ای خانوادگی). این بیماری در بین یهودیان، عرب ها، ارمنی ها شایع تر است. تظاهرات - درجه حرارت بالای 39 درجه سانتیگراد، درد در سر و عضلات، تعریق زیاد. التهاب غشای ریه ها، اندام های صفاقی، سینوویال وجود دارد. انحرافات مکرر در روان.
آمیلوئیدوز پیری
در افرادی که به سن 80 سالگی رسیده اند، آمیلوئید به صورت موضعی در بافت ها و اندام های مختلف رسوب می کند. این بیماری با سایر بیماری های مرتبط با افزایش سن همراه است. دو نوع آمیلوئیدوز پیری وجود دارد:
- مغزی یا مغزی. در پس زمینه بیماری آلزایمر ایجاد می شود. آمیلوئید آب در بافت مغز رسوب می کند.
- صمیمی می تواند بر بطن های قلب (زمانی که آمیلوئید از پروتئین جهش یافته خون ترانس تیرتین تشکیل می شود) و دهلیزها (زمانی که آمیلوئید از پپتید ناتریورتیک ترشح شده توسط سلول های قلب تشکیل می شود) تأثیر بگذارد. در هر دو مورد، آمیلوئیدها در بافت های ریه، پانکراس و طحال یافت می شوند.
برای تومورها
برخی از انواع تومورها بر تبدیل بدخیم سلول های اندام بیمار تأثیر می گذارند که در نتیجه پروتئین فیبریلار تولید می کنند. در این حالت آمیلوئیدوز به صورت موضعی در بافت اندام تحت تأثیر تومور ایجاد می شود. علل تحریک آمیلوئیدوز در تومورها:
- تومور مدولاری غده تیروئید. سرطان از سلول های C در غده تیروئید ایجاد می شود که وضعیت عادیمسئول تولید کلسی توسین است. هنگامی که سنتز کلسی توسین مختل می شود، قطعات آن بخشی از آمیلوئید AE می شوند.
- سرطان جزایر تیروئید. جزایر تجمع سلولهایی هستند که مسئول تولید هورمونها هستند - گلوکاگون، انسولین، سوماتوستاتین، و غیره. تخریب بدخیم سلولها باعث آزاد شدن پروتئین فیبریلار میشود که متعاقباً به آمیلوئید تبدیل میشود.
آمیلوئیدوز در همودیالیز
 همودیالیز یک روش نجات دهنده برای بیمارانی است که کلیه های آنها قادر به پاکسازی خون از سموم نیستند. محصولات جانبیمتابولیسم همودیالیز را به کسانی که نارسایی کلیوی دارند (حاد، مزمن) اختصاص دهید.
همودیالیز یک روش نجات دهنده برای بیمارانی است که کلیه های آنها قادر به پاکسازی خون از سموم نیستند. محصولات جانبیمتابولیسم همودیالیز را به کسانی که نارسایی کلیوی دارند (حاد، مزمن) اختصاص دهید.
ماهیت این روش عبور خون از طریق دستگاهی است که مواد مضر را از آن خارج می کند، بازگشت خون تصفیه شده به بدن بیمار.
در طی دیالیز، میکروگلوبولین B2 نمی تواند از بدن خارج شود، و اگر بیمار مدت زمان طولانیاین پروتئین که مجبور به همودیالیز می شود، در مقادیر بیش از حد در بدن تجمع می یابد. به نوکلئوپروتئین های پلاسما متصل می شود و در آن ته نشین می شود بدن های مختلف، پایه آمیلوئید می شود.
علائم آمیلوئیدوز
با توجه به اینکه بیماری می تواند به هر عضو یا بافتی سرایت کند، علائم متفاوت خواهد بود. اشکال گوناگونبیماری ها در ابتدای دوره با آسیب و اختلال در عملکرد یک اندام در بدن انسان مشخص می شوند.
با گذشت زمان، بیماری (اگر آمیلوئیدوز موضعی نباشد) پیشرفت می کند و سایر اندام ها و بافت ها را تحت تأثیر قرار می دهد. تظاهرات آمیلوئیدوز را می توان در کلیه ها، کبد، قلب و غدد فوق کلیوی، طحال، دستگاه گوارش و سیستم عصبی، مفاصل، ماهیچه ها و پوست مشاهده کرد. انواع بیماری در زیر به تفصیل توضیح داده شده است.
آسیب کلیه
آمیلوئیدوز کلیه بیشتر در نظر گرفته می شود بیماری خطرناکدر مقایسه با سایر اندام ها تصویر بالینی آمیلوئیدوز کلیه به مرحله بستگی دارد. در مجموع، 4 مورد از آنها وجود دارد - نهفته، نفروتیک، آزوتمیک، پروتئینوریک.
در مرحله نهفته، آمیلوئیدوز کلیه عملاً علائمی را نشان نمی دهد. اگر این شکل ثانویه باشد، بیمار علائم بیماری زمینه ای را احساس می کند. تنها سال ها بعد، آسیب کلیه به طور علامتی ظاهر می شود.
در مرحله پروتئینوری، آمیلوئیدوز کلیه ها 10 سال یا بیشتر طول می کشد. در این زمان آمیلوئیدوز به تدریج در عروق، فضای بین سلولی و گلومرول های کلیه رسوب می کند. به همین دلیل، نفرون هایی که تشکیل ادرار را فراهم می کنند فشرده می شوند، آتروفی می شوند و می میرند. یکپارچگی فیلتر کلیه که به طور معمول اجازه عبور پروتئین های مولکولی بزرگ و سلول های خونی را نمی دهد، شکسته می شود. پس از آن، پروتئین ها از طریق ادرار دفع می شوند. در این مرحله، مشکوک شدن به آمیلوئیدوز کلیه ها دشوار است، زیرا عملکرد دفعی آن مختل نمی شود. شما می توانید مشکل را در نتایج آزمایشات آزمایشگاهی پیدا کنید.
آمیلوئیدوز کلیه در مرحله نفروتیک با تخریب بیشتر فیلتر کلیه آشکار می شود. به همین دلیل، مقدار زیادی پروتئین در ادرار از بین می رود، غلظت آن در خون کاهش می یابد. پروتئین ها بخشی از فرآیند نگهداری خون در رگ ها هستند. با کاهش غلظت پروتئین ها، مایع وارد بافت ها می شود، تورم در هر زمانی از روز، صرف نظر از موقعیت بدن، رخ می دهد. علاوه بر این، آمیلوئیدوز کلیه ها پیشرفت می کند، ادم به شدت مشخص می شود. مایع در صفاق، حفره پلور، کیسه قلب تجمع می یابد. این مرحله 4-6 سال طول می کشد.
در مرحله آزوتمیک از حجم کل بافت کلیهفقط 25 درصد کار می کنند. این برای حذف سموم مضر، اوره کافی نیست و بنابراین غلظت آنها در حال افزایش است. تصویر بالینی نارسایی کلیه در موارد زیر آشکار می شود:
- اختلال در ادرار به جای 800 میلی لیتر تجویز شده در روز، بیمار کمتر از 50 میلی لیتر ادرار دفع می کند.
- سلامتی بدتر می شود، ضعف، خستگی ظاهر می شود.
- هضم مختل می شود، اشتها از بین می رود، حالت تهوع و استفراغ رخ می دهد، خشکی دهان با بوی نامطبوع همراه است.
- پوست رنگ پریده، خشک، دائما خارش می شود.
- سیستم قلبی عروقی رنج می برد، که باعث آریتمی، افزایش فشار خون، افزایش عضله قلب می شود.
- مغز تحت تمرکز بالا اسید اوریکآسیب دیده، بی خوابی و اختلال حافظه، تحریک پذیری، زوال ذهنی وجود دارد.
- کاهش هموگلوبین و گلبول های قرمز منجر به کم خونی می شود.
آسیب کبدی
آمیلوئیدوز سیستمیک اغلب با آسیب کبدی آشکار می شود. رسوبات آمیلوئید بر مجاری صفراوی، رگ های خونی و سلول های کبدی فشار وارد می کند و در نتیجه عملکرد اندام ها را مختل می کند. برجسته کردن سندرم های آمیلوئیدوز، نشان دهنده افزایش کبد است که در لمس احساس می شود.
سطح کبد صاف می ماند، هیچ دردی وجود ندارد. در صورت دوره طولانی بیماری، نارسایی کبد به ندرت ایجاد می شود که با توانایی های بازسازی اندام همراه است.
آمیلوئیدوز کبدی با علائم زیر ظاهر می شود:
- بزرگ شدن کبد.
- فشار خون پورتال به طور معمول، خون از اندام های داخلی وارد کبد می شود و در آنجا پاک می شود و سپس به جریان خون باز می گردد. هنگام فشردن رگ های کبد با آمیلوئید، فشار در وریدهای اندام های داخلی افزایش می یابد. در نتیجه، تورم در پاها، اسهال همراه با خون، خونریزی در دستگاه گوارش وجود دارد.
- زردی نادر است، فقط در صورت فشرده شدن مجاری صفراوی توسط رسوبات آمیلوئید. اگر این دلیل باشد، خارش با زردی همراه خواهد بود.
نارسایی قلبی
آمیلوئیدوز قلب به شکل اولیه و سایر اشکال طبیعت ارثی ایجاد می شود. در نتیجه رسوب آمیلوئید در میوکارد و غشاهای قلب، گردش خون مختل می شود. سلول های ماهیچه ایدارد می میرد.
علائم بیماری:
- آریتمی؛
- کاردیومیوپاتی محدود کننده؛
- نارسایی قلبی.
آریتمی در پس زمینه رسوب آمیلوئید در عضله قلب رخ می دهد که باعث اختلال در هدایت یک تکانه عصبی می شود. در نتیجه، اتاق های قلب به طور ناهموار منقبض می شوند، آریتمی ظاهر می شود. بیمار احساس سرگیجه می کند، غش مشاهده می شود. به دلیل اختلال در خون رسانی به مغز، یک عاقبت کشنده ممکن است.
کاردیومیوپاتی محدود کننده در پس زمینه رسوب آمیلوئید در میوکارد رخ می دهد. در نتیجه، عضله قلب ضخیم می شود، کمتر قابل انبساط می شود، که منجر به عملکرد ضعیف حفره های قلب می شود. تصویر بالینی بیماری خستگی، تنگی نفس، کاهش شدید فشار خون هنگام تغییر است موقعیت افقیبه موقعیت عمودی
با نارسایی قلبی، گردش خون در بدن مختل می شود. این با تورم، تنگی نفس آشکار می شود. نارسایی قلبی در آمیلوئیدوز قابل درمان استاندارد برای بیماری های قلبی عروقی نیست. این بیماری به سرعت پیشرفت می کند و در عرض چند ماه منجر به مرگ می شود.
آسیب به غدد فوق کلیوی و طحال
 غدد فوق کلیوی غددی هستند که در هر کلیه قرار دارند و مسئول ترشح هورمون ها هستند. آمیلوئیدوز با توقف سنتز هورمون، عملکرد اندام ها را مختل می کند. اگر آمیلوئید در طحال رسوب کند، اندازه اندام افزایش می یابد که در لمس قابل توجه است.
غدد فوق کلیوی غددی هستند که در هر کلیه قرار دارند و مسئول ترشح هورمون ها هستند. آمیلوئیدوز با توقف سنتز هورمون، عملکرد اندام ها را مختل می کند. اگر آمیلوئید در طحال رسوب کند، اندازه اندام افزایش می یابد که در لمس قابل توجه است.
به طور معمول، طحال سلول های تغییر شکل یافته جریان خون را که در ساختار آن گیر کرده اند، از بین می برد. رسوب آمیلوئید در طحال باعث می شود گلبول های قرمز سالم، پلاکت ها و گلبول های سفید خون گیر کنند.
در نتیجه، کم خونی (ضعف عمومی، رنگ پریدگی پوست، تنگی نفس)، ترومبوسیتوپنی (خونریزی بینی، خونریزی های پوستی)، لکوپنی (حساسیت به عفونت) ایجاد می شود.
ضایعه گوارشی
آمیلوئیدوز روده ای می تواند تعمیم یابد، زمانی که جذب مواد مغذی مختل می شود، و زمانی که تجمع آمیلوئید شبیه یک تومور است، موضعی است. در حالت اول علائمی مانند اسهال، کاهش وزن، ضعف، اختلالات روانی، کم خونی ظاهر می شود. در مورد دوم، بیماری با یبوست، درد شکم، نفخ مشخص می شود.
آسیب مفاصل و عضلات
آمیلوئید ابتدا ضربه می زند مفاصل کوچکروی پاها، دست ها، با پیشرفت بیماری، در آرنج و زانو می نشیند. این بیماری با درد در هنگام حرکت، تورم بافت و قرمزی پوست، تب در ناحیه آسیب دیده، اختلال در عملکرد مفصل مشخص می شود.
آمیلوئید بدون ایجاد اختلال در ساختار عضلانی و بدون ظاهر شدن به مدت طولانی به صورت نامحسوس در بافت همبند رسوب می کند. با گذشت زمان، سلول های بافت ماهیچه ای فشرده می شوند، خون رسانی به آنها مختل می شود و می میرند. این بیماری با ضعف عضلانی، درد، سفت شدن و هیپرتروفی عضلانی مشخص می شود.
تشخیص آمیلوئیدوز
تشخیصی مانند آمیلوئیدوز می تواند توسط پزشکان تخصص های مختلف - روماتولوژیست، متخصص قلب و اورولوژی، متخصص مغز و اعصاب، متخصص پوست و غیره مشکوک باشد. بنابراین، تشخیص آمیلوئیدوز باید بر اساس ارزیابی جامع تاریخ باشد علائم بالینی، آزمایشگاه و معاینه ابزاری. برای بررسی وضعیت اندام ها، نوار قلب، اشعه ایکس مری، آندوسکوپی، سیگموئیدوسکوپی تجویز می شود. اگر مشکوک به آمیلوئیدوز کلیه ها باشد، تشخیص لزوماً شامل سونوگرافی حفره شکمی است.
درمان آمیلوئیدوز
اگرچه متفاوت هستند بیماری جدیآمیلوئیدوز پیش آگهی بدی دارد. واقعیت این است که در مراحل اولیه تشخیص بیماری ممکن نیست و تظاهرات بالینی آن سال ها پس از شروع بیماری قابل توجه است. با چنین تشخیصی مانند آمیلوئیدوز کلیه، درمان فقط حمایتی است، زیرا اقدامات درمانی موثر نیستند.
در اولین شک به وجود بیماری بستری در نفرولوژی برای معاینه ضروری است. سیستم تناسلی ادراری، از آنجایی که آسیب کلیه بیشتر در نظر گرفته می شود تجلی خطرناک. متخصصان دیگری نیز برای بررسی وجود آسیب به سایر اندام ها درگیر هستند.
اگر تشخیص نقض جدی در عملکرد حیاتی را نشان نداد اندام های مهم، درمان آمیلوئیدوز را می توان در خانه انجام داد، جایی که بیمار باید به شدت از تمام دستورات پزشک پیروی کند. درمان ممکن است شامل دارو، رژیم غذایی، دیالیز و پیوند اعضا باشد.
"آمیلوئیدوز" اصطلاحی است که گروهی از بیماری ها را که بسیار متنوع هستند، متحد می کند. تظاهرات بالینیو با رسوب خارج سلولی پروتئین های فیبریلار پاتولوژیک نامحلول در اندام ها و بافت ها مشخص می شوند. این آسیب شناسی اولین بار در قرن هفدهم توصیف شد. بونت - طحال ساگو در بیمار مبتلا به آبسه کبدی. در اواسط قرن نوزدهم. ویرکو از اصطلاح گیاه شناسی "آمیلوئید" (از واژه یونانی آمیلون، نشاسته) برای توصیف مواد خارج سلولی یافت شده در کبد در کالبد شکافی استفاده کرد، زیرا معتقد بود که از نظر ساختار شبیه به نشاسته است. پس از آن، ماهیت پروتئینی رسوبات مشخص شد، اما اصطلاح "آمیلوئید" تا به امروز حفظ شده است.
در دهه 20. در قرن بیستم، بنهولد پیشنهاد کرد که آمیلوئید را با رنگ قرمز کنگو رنگ آمیزی کند، سپس اثر شکست مضاعف در نور قطبی شده کشف شد - تغییر از قرمز آجری به سبز سیبی. در سال 1959، کوهن و کالکینز ساختار فیبریل آمیلوئید را با استفاده از میکروسکوپ الکترونی ایجاد کردند.
مفاهیم بالینی آمیلوئیدوز نیز دستخوش تکامل شده است: روکیتانسکی در سال 1842 ارتباطی بین "بیماری چربی" و سل، سیفلیس و ریکتسیوز ایجاد کرد. ویلکس در سال 1856 " اندام های چربی " را در یک بیمار که هیچ بیماری همزمان نداشت توصیف کرد. اتکینسون در سال 1937 آمیلوئیدوز را در بیماران مولتیپل میلوما کشف کرد. اشکال پیری (Soika، 1876) و ارثی (Andrade، 1952) بیماری شناسایی شد، آمیلوئیدوز به ژنتیک، اولیه و ثانویه تقسیم شد و در نهایت، در سال 1993، طبقه بندی WHO بر اساس ویژگی پروتئین آمیلوئید فیبریلار اصلی به تصویب رسید.
در کشور ما، E. M. Tareev، I. E. Tareeva، V. V. Serov سهم بزرگی در توسعه ایده ها در مورد آمیلوئیدوز داشته است. نقش بزرگی در مطالعه انواع اولیه و ژنتیکی آمیلوئیدوز و بیماری های دوره ای متعلق به O. M. Vinogradova است که تک نگاری های او که در سال های 1973 و 1980 منتشر شده اند، امروزه ارتباط خود را از دست نداده اند.
در حال حاضر آمیلوئیدوز از نظر بالینی به سیستمیک و اشکال محلی. در بین اشکال سیستمیک، بسته به ترکیب رسوبات فیبریلار، چهار نوع وجود دارد ( ).
اشکال محلی آمیلوئیدوز در حال حاضر شامل بیماری آلزایمر (A-بتا، فیبریلها شامل پروتئین بتا است که در مغز رسوب میکند)، آمیلوئیدوز جزایر پانکراس، احتمالاً ارتباط پاتوژنتیکی با دیابت نوع 2، آمیلوئیدوز که در تومورهای غدد درون ریز، تومورهای آمیلوئیدی پوست و سایر گونههای نازوفادری رخ میدهد.
آمیلوئیدوز AL
ایجاد AL-آمیلوئیدوز با مولتیپل میلوما، بیماری والدنستروم، لنفوم های سلول Bو ممکن است در آمیلوئیدوز اولیه ایدیوپاتیک باشد. همه این گزینه ها ترکیب شده اند پاتوژنز رایجتشخیص آمیلوئیدوز اولیه بدلیل فقدان آن سخت ترین است نشانه های آشکار بیماری هماتولوژیکبنابراین، ارزش دارد که در این فرم به تفصیل صحبت کنیم.
در آمیلوئیدوز اولیه، دیسکرازی خوش خیم سلول های پلاسما، مرتبط است مولتیپل میلوماکلون های غیر طبیعی سلول های پلاسما مغز استخوان، ایمونوگلوبولین های آمیلوئیدوژن تولید می کنند. برخی از اسیدهای آمینه در نواحی متغیر زنجیره های سبک این ایمونوگلوبولین ها موقعیت غیرعادی را اشغال می کنند که منجر به ناپایداری آنها و تمایل به فیبریلوژنز می شود. در بیماران مبتلا به آمیلوئیدوز اولیه، محتوای سلول های پلاسما در مغز استخوان به 5-10٪ افزایش می یابد (به طور معمول کمتر از 4٪، با میلوم متعدد - بیش از 12٪)، و ایزوتیپ خاصی از زنجیره های سبک ایمونوگلوبولین تولید می کنند که در رنگ آمیزی ایمونوهیستوشیمی غالب است. زنجیره های سبک مونوکلونال آزاد ایزوتیپ لامبدا یا (به طور معمول) کاپا غالب در خون و ادرار شناسایی می شوند، اما محتوای آنها کمتر از مولتیپل میلوما است.
تصویر بالینی آمیلوئیدوز اولیه متنوع است و با درگیری غالب اندام های خاص در فرآیند پاتولوژیک - قلب، کلیه ها، سیستم عصبی، دستگاه گوارش، کبد و غیره تعیین می شود. اولین علائم ضعف و کاهش وزن است، اما در این مرحله، قبل از ظهور علائم اندام، تشخیص بسیار نادر است.
شایع ترین اندام های هدف در آمیلوئیدوز AL کلیه ها و قلب هستند. آسیب کلیه با سندرم نفروتیک آشکار می شود، مداوم و با شروع نارسایی مزمن کلیه، هماچوری و فشار خون شریانی معمول نیست.
هنگامی که آمیلوئید در میوکارد رسوب می کند، تخلفات مختلفریتم، نارسایی پیشرونده قلب، که ممکن است با تغییرات بدون علامت در ECG به شکل کاهش ولتاژ دندان ها قبل از آن باشد. اکوکاردیوگرافی ضخیم شدن متحدالمرکز دیوارههای بطن چپ و راست، کاهش حجم حفرههای قلب، کاهش متوسط کسر جهشی و اختلال دیاستولیک میوکارد بطن چپ را نشان میدهد.
اغلب علائم درگیری سیستم عصبی - خودمختار، به شکل وجود دارد افت فشار خون ارتواستاتیک، و محیطی - به شکل اختلالات حساسیت. در سالهای اخیر، ضایعات سیستم عصبی مرکزی نیز توصیف شده است، اگرچه قبلاً اعتقاد بر این بود که آنها مشخصه آمیلوئیدوز اولیه نیستند.
علائم سوء هاضمه (احساس پری، یبوست، اسهال) و سندرم سوء جذب می تواند هم در اثر آسیب به سیستم عصبی خودمختار و هم آمیلوئیدوز دستگاه گوارش ایجاد شود. هپاتومگالی بسیار مشخص است که ماهیت آن را باید بین احتقان ناشی از نارسایی قلبی و آسیب کبدی آمیلوئید افتراق داد. مورد دوم با افزایش سطح آلکالین فسفاتاز در سرم خون تأیید می شود. طحال اغلب تحت تأثیر قرار می گیرد، اما طحال همیشه پیدا و بزرگ نیست اهمیت بالینیندارد.
ماکروگلوسیا، یک علامت کلاسیک آمیلوئیدوز اولیه، در 20٪ از بیماران رخ می دهد؛ نفوذ بافت نرم می تواند منجر به آتروفی عضلانی و پوست، دیستروفی ناخن، آلوپسی، و ظهور تشکیلات تومور مانند - آمیلوئید شود.
آسیب عروقی کمتر شایع است که علائم آن پورپورای اطراف چشم - "چشم های راکون" و اکیموز است. ممکن است خونریزی، از جمله خونریزی مثانه، ناشی از تغییرات در دیواره عروق و نقض سیستم انعقادی، در درجه اول کمبود فاکتور X، که به آمیلوئید متصل می شود، ایجاد شود. مرسوم است که مشخصه ترومبوسیتوز آمیلوئیدوز را با کمبود فاکتورهای انعقادی توضیح دهیم.
آمیلوئیدوز ریوی اغلب فقط در کالبد شکافی یافت می شود. با این حال، در برخی موارد، تنگی نفس، هموپتیزی و هیدروتوراکس می تواند نه تنها به دلیل نارسایی احتقانی قلب و سندرم نفروتیک، بلکه به دلیل بیماری ریه آمیلوئید نیز ایجاد شود. رسوب آمیلوئید در آلوئول ها و ایجاد آمیلوئیدوم ریوی امکان پذیر است. از طریق رادیوگرافی، تغییرات مش و گرهی در بافت ریه قابل تشخیص است.
درگیری آدرنال می تواند منجر به نارسایی آدرنال شود که اغلب ناشناخته می ماند زیرا افت فشار خون و هیپوناترمی به عنوان علائم نارسایی قلبی و آسیب سیستم عصبی اتونومیک دیده می شود. در 10-20٪ از بیماران، کم کاری تیروئید ممکن است به عنوان تظاهرات آسیب تیروئید رخ دهد، اغلب افزایش در غدد بزاقی زیر فکی وجود دارد.
تشخیص آمیلوئیدوز اولیه، علاوه بر ویژگی های بالینی نشان داده شده، که ممکن است در آمیلوئیدوز ثانویه مشابه باشد، بر اساس تعدادی از داده های آزمایشگاهی است. در 85 درصد بیماران، ایمونوالکتروفورز پروتئین های سرم خون و ادرار، ایمونوگلوبولین های مونوکلونال را نشان می دهد. در مطالعات معمول، همان ایمونوگلوبولین های مونوکلونال در ادرار به شکل پروتئین Bence-Jones یافت می شود. بیوپسی مغز استخوان اجازه می دهد تشخیص های افتراقیبا مولتیپل میلوما، و همچنین برای تشخیص افزایش متوسط در تعداد سلول های پلاسما و مونوکلونالیته آنها با رنگ آمیزی ایمونوهیستوشیمی.
با این حال، حتی ترکیبی از یک تصویر بالینی مشخص و وجود سلولها و پروتئینهای پلاسما مونوکلونال هنوز برای تأیید تشخیص آمیلوئیدوز اولیه کافی نیست. داده های بیوپسی در اینجا نقش تعیین کننده ای دارند. کمترین تهاجم، آسپیراسیون بافت چربی زیر جلدی قدامی است دیواره شکم، دادن 80-90٪ نتایج مثبتبا AL- آمیلوئیدوز (در کشور ما این روش هنوز کاربرد پیدا نکرده است). بیوپسی از لثه و مخاط رکتوم ارزش تشخیصی خاصی دارد، اما درصد نتایج مثبت به طور گسترده ای بسته به مرحله فرآیند متفاوت است، بنابراین توصیه می شود بیوپسی یکی از اندام های آسیب دیده - کلیه، کبد، قلب، انجام شود که تقریباً 100٪ نتایج مثبت در آمیلوئیدوز نوع AL می دهد.
اول از همه، مواد بیوپسی با رنگ قرمز کنگو رنگ آمیزی می شود. اگر کنگوفیلی مواد مورد مطالعه تشخیص داده شود، لازم است آن را در نور پلاریزه مطالعه کنید، اثر انکسار دوگانه فقط برای آمیلوئید مشخص است، سایر مواد کنگوفیلیک رنگ سبز سیبی به دست نمی آورند. پس از آن، تایپ آمیلوئید مطلوب است. دقیق ترین روش ایمونوهیستوشیمی با استفاده از آنتی بادی های مونوکلونال به پروتئین های پیش ساز آمیلوئید است. اما در حال حاضر در کشور ما عملاً در دسترس نیست. بنابراین، رنگ آمیزی با محلول های گوانیدین قلیایی یا پرمنگنات پتاسیم برای تشخیص استفاده می شود، که امکان تعیین نوع رسوبات فیبریل را، هرچند غیرمستقیم، فراهم می کند.
پیش آگهی آمیلوئیدوز اولیه بدتر از سایر اشکال این بیماری است. مدت زمان متوسطعمر از دو سال تجاوز نمی کند، در صورت وجود بیماری قلبی یا ضایعات چند سیستمی بدون درمان، بیماران در عرض چند ماه می میرند. اکثر علل شایعمرگ و میر عبارتند از نارسایی قلبی و کلیه، سپسیس، عوارض عروقی و کاشکسی. شباهت پاتوژنتیک با مولتیپل میلوما به ما امکان می دهد در طول شیمی درمانی، که برای سرکوب سلول های پلاسما مونوکلونال انجام می شود، روی مهار پیشرفت بیماری حساب کنیم. چندین رژیم درمانی وجود دارد ().
استفاده از شیمی درمانی در صورت موفقیت آمیز بودن درمان می تواند طول عمر بیماران را برای مدت 10 تا 18 ماه افزایش دهد. اما اثربخشی درمان پایین است، به ویژه به این دلیل که در بسیاری از موارد پیشرفت بیماری منجر به مرگ بیماران قبل از اتمام دوره درمان و همچنین به دلیل ایجاد سیتوپنی می شود. عوارض عفونی، آریتمی های کشنده در درمان دوزهای فوق العاده بالای دگزازون. استفاده از دوزهای بالای ملفولان با پیوند سلولهای بنیادی اتولوگ امکان دستیابی به بهبودی را در بیش از 50 درصد موارد فراهم میکند، اما استفاده از این روش با توجه به شدت بیماری، سن بیماران، محدود میشود. اختلالات عملکردیاز قلب و کلیه ها در بسیاری از موارد، تنها درمان حمایتی علامتی امکان پذیر است.
آمیلوئیدوز AA
توسعه AA-آمیلوئیدوز در طول فرآیندهای التهابی مزمن رخ می دهد، پیش سازهای AA-آمیلوئید پروتئین های مرحله حاد سرم، α-گلوبولین های تولید شده توسط سلول ها هستند. انواع متفاوتعمدتا نوتروفیل ها و فیبروبلاست ها. آمیلوئیدوز ثانویه در آرتریت روماتوئید، بیماری Bechterew، آرتریت پسوریاتیک، تومورهای مختلف، بیماری هوچکین، کولیت اولسراتیو و بیماری کرون، بیماری های دوره ای (تب مدیترانه ای خانوادگی) و همچنین سل، استئومیلیت، برونشکتازی ایجاد می شود.
ویژگی های بالینی مشخص آمیلوئیدوز AA آسیب کلیه در اکثر بیماران و همچنین آسیب نسبتاً نادر به کبد و / یا طحال (حدود 10٪) و قلب (فقط با اکوکاردیوگرافی تشخیص داده می شود). ماکروگلوسیا برای آمیلوئیدوز ثانویه معمول نیست. تشخیص بر اساس ترکیبی از آمیلوئیدوز کلیه و یک بیماری التهابی مزمن است که با رنگ آمیزی ایمونوهیستوشیمیایی مواد بیوپسی تایید می شود؛ در کشور ما از روش های رنگ آمیزی غیرمستقیم که قبلا ذکر شد استفاده می شود.
پیش آگهی تا حد زیادی به ماهیت بیماری زمینه ای بستگی دارد؛ در یک دوره طبیعی، یک سوم بیماران 5 سال پس از تشخیص پروتئینوری دچار نارسایی کلیوی می شوند. با بیماری دوره ای، میزان بقای پنج ساله 25٪ است.
درمان مبتنی بر سرکوب تمرکز - منبع تولید پروتئین های پیش ساز سرم است. برداشتن تومورها، سکوسترکتومی، برداشتن روده، درمان سل، کاهش فعالیت آرتریت روماتوئید (با استفاده از سیتواستاتیک) منجر به توقف پیشرفت آمیلوئیدوز و گاهی اوقات به پیشرفت معکوس تظاهرات بالینی، به ویژه سندرم نفروتیک می شود.
استفاده از کلشی سین در بیماری های دوره ای روش انتخابی است، اثربخشی آن ثابت شده است، درمان از ایجاد آمیلوئیدوز جلوگیری می کند و پیشرفت آن را کند می کند. در سایر اشکال آمیلوئیدوز ثانویه، اثربخشی کلشی سین تایید نشده است.
اشکال سالخورده و ارثی آمیلوئیدوز سیستمیک، و همچنین اشکال موضعی، نادر هستند، آمیلوئیدوز دیالیز به خوبی برای متخصصان شناخته شده است، در عمل عمومی تقریبا هرگز با آن مواجه نمی شود.
درمان علامتی به نوع آمیلوئیدوز بستگی ندارد، بلکه به اندام های هدف آسیب دیده بستگی دارد. ).
آمیلوئیدوز، به ویژه اولیه، یک آسیب شناسی نادر در نظر گرفته می شود، اما در واقعیت آنقدر نادر نیست که تشخیص آن دشوار است. تشخیص کافی نه تنها نیازمند آگاهی از کلینیک و پاتوژنز این بیماری، بلکه در دسترس بودن قابلیت های تشخیصی خاص است. برای نشان دادن این نکته، ما داده های خود را ارائه می کنیم ( ). در بخش نفرولوژی بیمارستان بالینی شهر مسکو به نام S.P. Botkin در سال 1993-2003. 88 بیمار با تشخیص آمیلوئیدوز مشاهده شدند.
تشخیص مورفولوژیکی در همه بیماران مبتلا به AL-آمیلوئیدوز، آمیلوئیدوز پیر و نامشخص، و در 30 بیمار مبتلا به آمیلوئیدوز ثانویه - در مجموع 53 مورد تأیید شد. بیوپسی کلیه در 12 بیمار، بیوپسی کبد در 2 بیمار، بیوپسی روده در 8 بیمار، لثه در 12 مورد انجام شد و در 19 مورد تشخیص با بررسی مورفولوژیکی مواد مقطعی تایید شد.
در اغلب موارد، تشخیص آمیلوئیدوز برای اولین بار در نتیجه معاینه در بخش نفرولوژی مشخص شد. ما تشخیص های ارجاعی و بالینی را در بین بیماران مبتلا به AL-آمیلوئیدوز مقایسه کردیم. ).
تنها در دو مورد از 20 مورد (10%)، تشخیص ارجاع آمیلوئیدوز اولیه بود و در یکی از این بیماران در کلینیک درمان و بیماری های شغلی MMA و در دیگری در کلینیک خارجی انجام شد.
تمام بیمارانی که مبتلا به مولتیپل میلوم با ایجاد AL-آمیلوئیدوز تشخیص داده شدند به بخش هماتولوژی منتقل شدند. از 11 بیمار مبتلا به آمیلوئیدوز اولیه، هفت بیمار شیمی درمانی با ترکیبی از ملفولان خوراکی و پردنیزولون را در دوره های متناوب دریافت کردند، چهار نفر از آنها همراه با درمان دیالیز و یک بیمار دیگر فقط دیالیز و دیالیز دریافت کردند. درمان علامتی. از این بیماران، پنج بیمار طی دو هفته تا دو سال پس از درمان فوت کردند (همه با نارسایی کلیوی و آسیب چند عضوی)، یک بیمار تحت دیالیز، یک بیمار برای پیوند سلول های بنیادی اتولوگ ارجاع شد و یک بیمار همچنان تحت درمان است. در یک بیمار، شیمی درمانی به دلیل وجود زخم معده طولانی مدت بدون اسکار به تعویق افتاد و دو بیمار دیگر از درمان خودداری کردند.
در بین بیماران مبتلا به آمیلوئیدوز ثانویه در مطالعه ما، بیماران مبتلا به آرتریت روماتوئید غالب بودند، در رتبه دوم از بین علل، استئومیلیت مزمن و آرتریت پسوریاتیک و سایر بیماری ها کمتر شایع بودند. ).
درمان آرتریت روماتوئید و آرتریت پسوریاتیک با استفاده از سیتواستاتیک (متاتروکسات، آزاتیوپرین) انجام شد، اگرچه در بسیاری از موارد به دلیل وجود CRF و بیماریهای همراه، امکانات درمانی محدود بود. بیماران مبتلا به استئومیلیت مزمن به بخش های جراحی چرکی ارجاع داده شدند. بیماران مبتلا به بیماری Bechterew و بیماری کرون تحت درمان اختصاصی قرار گرفتند، بیماران مبتلا به COPD و سل نیز به بیمارستان های تخصصی ارجاع شدند. یکی از بیماران مبتلا به تومور معده با موفقیت مورد عمل قرار گرفت و در طی چهار سال مشاهده، سندرم نفروتیک به تدریج پسرفت کرد، در موارد دیگر تومورها، شیوع این روند فقط به درمان علامتی اجازه داد، یک بیمار مبتلا به لنفوگرانولوماتوز در بیمارستان بستری شد. حالت ترمینال. مرگ و میر در میان بیماران مبتلا به آمیلوئیدوز ثانویه 38 درصد بود (به دلیل بیمارانی که ضایعات پیشرفته در زمان تشخیص داشتند). تمام بیماران مبتلا به بیماری دوره ای درمان با کلشیسین دریافت کردند.
ویژگی های تشخیص و استفاده از روش های نوین درمان آمیلوئیدوز اولیه را می توان با مثال زیر نشان داد: بیمار K.، 46 ساله، برای اولین بار در اواخر اکتبر 2002 با شکایت تورم در پاها، تپش قلب، آمنوره در بیمارستان بستری شد. در تاریخ - سرماخوردگیآپاندکتومی، دو زایمان سریع طبیعی، نشانه های بیماری کلیوی، بدون بیماری مزمن. در آوریل 2002 نقل مکان کرد پنومونی حاددر لوب فوقانی ریه راست، به صورت سرپایی درمان شد، تزریق آباکتال، لینکومایسین دریافت کرد. با توجه به محلی شدن ذات الریه، او در یک داروخانه سل معاینه شد، تشخیص سل منتفی شد. در اوایل ژوئن، برای اولین بار، ادم روی پاها ظاهر شد، که او مورد بررسی قرار نگرفت. تورم از طریق مدت کوتاهیخود منحل شد، سپس از سر گرفته شد. بیمار در یک بیمارستان درمانی بستری شد، در معاینه پروتئینوری تا 1.65٪، هیپوپروتئینمی (کل پروتئین سرم 52 گرم در لیتر)، فشار خون طبیعی (120/80 میلی متر جیوه)، رسوب ادرار بدون تغییر، کراتینین پلاسما نیز در محدوده طبیعی مشاهده شد. تشخیص "گلومرولونفریت حاد" مشخص شد، درمان با آمپی سیلین، کیمز، هپارین، تری آمپور انجام شد، برداشتن لوزه انجام شد. پروتئینوری ادامه داشت، ادم به تدریج افزایش یافت و بنابراین برای معاینه و درمان بیشتر، بیمار با تشخیص گلومرولونفریت مزمن به بیمارستان اعزام شد. S. P. Botkin.
در معاینه، پوست تمیز، رنگ طبیعی، آناسارکا، تورم عظیم، متراکم، آسیت مشخص شده، غدد لنفاوی محیطی بزرگ نشده است. BP 110/70 میلی متر جیوه. هنر، صداهای قلب صوتی، شفاف، ریتمیک، ضربان قلب 90 ضربه در دقیقه است، کبد و طحال بزرگ نمی شوند، دیورز تا 1000 میلی لیتر در روز است، مدفوع منظم، بدون ناخالصی های پاتولوژیک است. معاینه نشان داد سندرم نفروتیک - پروتئینوری 3 گرم در لیتر، رسوب ادرار کمی، هیپودیسپروتئینمی، هیپرلیپیدمی (کل پروتئین سرم 39 گرم در لیتر، آلبومین 12 گرم در لیتر، گلوبولین ها 7-30-15-19 درصد، α1 -α 2 -2-β-α-2-5-β-γ1، α1-α2-chole-β-5-γ-1 mml-β-5-γ. واحد)، تجزیه و تحلیل ادرار برای پروتئین Bence-Jones - واکنش منفی است، دفع روزانه 17-KS کاهش نمی یابد. آزمایش خون بالینی و سایر پارامترهای بیوشیمیایی در محدوده طبیعی است، کواگولوگرام - هیپرفیبرینوژنمی شدید، افزایش سطح RKFM. مطالعه ایمونوگلوبولین های خون: Ig-A - 0.35، Ig-M - 35.7 (دو هنجار)، Ig-G - 1.96 گرم در لیتر. اشعه ایکس از قفسه سینه، استخوان های جمجمه و لگن، سونوگرافی حفره شکمی، کلیه ها، غده تیروئید، ECHO-KG بدون آسیب شناسی، سونوگرافی لگن کوچک - علائم آدنومیوز بدن رحم، آندوسکوپی - ازوفاژیت رفلاکس، گاستریت مزمن. هنگامی که توسط یک نوروپاتولوژیست معاینه شد، هیچ آسیب شناسی یافت نشد؛ یک انکولوژیست ماستوپاتی فیبروکیستیک را تشخیص داد.
به منظور روشن شدن پیدایش سندرم نفروتیک تحت بیهوشی موضعی تحت هدایت سونوگرافی، بیوپسی با سوزن نازک از کلیه راست انجام شد که هیچ عارضه ای نداشت. در مطالعه بیوپسی در مزانژیوم گلومرول ها و در عروق خارج گلومرولی، رسوب آمیلوئید ذکر شده است. آمیلوئید تا 25 درصد از حلقه های عروقی گلومرولی را بارگیری می کند. مطالعه ایمونوهیستوشیمی لومینسانس خاصی را نشان نداد. هنگامی که آماده سازی با محلول قلیایی گوانیدین به مدت 2 ساعت درمان می شود، کنگوفیلی و خواص آنها در نور پلاریزه حفظ می شود، که برای AL-amyloidosis معمول است.
برای روشن شدن ماهیت AL-آمیلوئیدوز، مطالعه ایمونوشیمیایی خون و ادرار در آزمایشگاه ایمونوتست انجام شد. پاراپروتئینمی M-lambda با کاهش سطح ایمونوگلوبولین های پلی کلونال و پاراپروتئینوری نوع لامبدا بنس جونز در برابر پس زمینه پروتئینوری غیرانتخابی عظیم آشکار شد. بیمار توسط متخصص هماتولوژیست مشاوره شد، پیشنهاد شد که بیماری والدنستروم وجود دارد و بیوپسی ترفین مغز استخوان انجام شد. نتیجهگیری: در حفرههای مغز استخوان موجود، سلولهای هر سه جوانه خونسازی طبیعی و همچنین سلولهای لنفوئیدی که خوشهای تشکیل نمیدهند، قابل مشاهده است. تشخیص بیماری والدنستروم به دلیل عدم وجود انفیلتراسیون لنفاوی مغز استخوان، بزرگ شدن غدد لنفاوی و طحال و عدم وجود بستر تومور رد شد.
تشخیص آمیلوئیدوز اولیه با آسیب کلیه، سندرم نفروتیک، حفظ عملکرد کلیه ایجاد شد، هیچ نشانه ای از آسیب اندام دیگر مشاهده نشد. از ژانویه 2003، شیمی درمانی با ملفولان 16 میلی گرم در روز و پردنیزولون 100 میلی گرم در روز، دوره های چهار روزه هر شش هفته آغاز شد. درمان علامتی نیز انجام می شود: فوروزماید، وروشپیرون، آماده سازی پتاسیم، فاموتیدین، تزریق آلبومین. تا به امروز، پنج دوره شیمی درمانی با تحمل خوب انجام شده است، ادم کاهش یافته، پروتئینوری به 1.8 گرم در لیتر کاهش یافته است، شدت هیپودیسپروتئینمی اندکی کاهش یافته است (کل پروتئین 46 گرم در لیتر، آلبومین 18 گرم در لیتر، α2-گلوبولین ها 20 درصد). عملکرد کلیه دست نخورده باقی می ماند، کراتینین پلاسما 1.3 میلی گرم در دسی لیتر است، هیچ نشانه ای از آسیب به سایر اندام ها و سیستم ها در طول بررسی های دینامیکی کنترل مشاهده نشد.
این مورد به وضوح این واقعیت را نشان می دهد که بررسی های مورفولوژیکی، ایمونولوژیکی و ایمونوشیمیایی برای تشخیص آمیلوئیدوز ضروری است. بنابراین در بیمار ما بارزترین تشخیص بالینی «گلومرولونفریت مزمن» بود و در صورت عدم امکان انجام بیوپسی کلیه، به احتمال زیاد این تشخیص داده می شد. بدون نشانه های بالینی ماهیت سیستمیک بیماری، مزمن فرآیند التهابی، بیماری سیستم خونی، به استثنای افزایش سطح Ig-M، بیمار نداشت. و تنها داده های به دست آمده در مطالعه بیوپسی کلیه منجر به ترپانوبیوپسی مغز استخوان و یک مطالعه ایمونوشیمیایی شد که با هم تشخیص آمیلوئیدوز اولیه را قبل از ظهور آسیب سیستمیک ممکن کرد. درمان پاتوژنتیکشروع شد، اگرچه در پس زمینه یک سندرم نفروتیک قبلاً توسعه یافته، اما قبل از شروع نارسایی کلیوی و تنها با 25٪ از گلومرول ها با آمیلوئید بارگذاری شده است، که از نظر پیش آگهی نسبتاً مطلوب است.
در خاتمه متذکر می شویم که آمیلوئیدوز a بیماری جدیبا سطح بالامرگ و میر، که تشخیص آن بسیار دشوار است، با این حال، معاینه به موقع و با کیفیت بالای بیماران امکان تشخیص زودتر را فراهم می کند. قرار به موقعدرمان کافی، به نوبه خود، بهبود پیش آگهی را در این گروه از بیماران ممکن می سازد.
ادبیات
- Varshavsky V. A., Proskurneva E. P. اهمیت و روشهای تشخیص مورفولوژیکی آمیلوئیدوز در پزشکی مدرن// نفرولوژی عملی. - 1998. - 2:16-23.
- Vinogradova OM انواع اولیه و ژنتیکی آمیلوئیدوز. - M.: پزشکی، 1980.
- Zakharova E. V.، Khrykina A. V.، Proskurneva E. P.، Varshavsky V. A. موردی از آمیلوئیدوز اولیه: مشکلات در تشخیص و درمان // نفرولوژی و دیالیز. - 2002. - 1:54-61.
- Rameev VV ویژگیهای آسیب کلیه در AA و AL-amyloidosis: diss... cand. عسل. علوم. - م.، 2003.
- Kozlovskaya L. V.، Varshavsky V. A.، Chegaeva T. V. و همکاران آمیلوئیدوز: ظاهر مدرندر مورد مشکل // نفرولوژی عملی. - 1998. - 2:24-26.
- رادنی اچ.، ریموند ال سی و اسکینر ام. // آمیلوئیدوزهای سیستمیک؛ مجله پزشکی نیوانگلند، 1997. 337:898-909.
- Dhodapkar M.V.، Jagannath S.، Vesole d. و همکاران // درمان AL-آمیلوئیدوز با دگزامتازون به علاوه آلفا اینترفرون / لنفوم Leuc. 1997. - 27(3-4):351-365
- Gertz M.A.، Lacy M.Q.، Lust J.A. و همه // فاز دوم کارآزمایی دگزامتازون با دوز بالا برای آمیلوئیدوز زنجیره سبک ایمونوگلوبولین درمان شده قبلی. ام جی هماتول، 1999، 61 (2): 115-119.
- Gertz M.A.، Lacy M.، Q.، Lust J.A. و همکاران // کارآزمایی فاز دوم دگزامتازون با دوز بالا در بیماران درمان نشده مبتلا به آمیلوئیدوز سیستنیک اولیه. Med Oncol 1999.-16(2):104-109
- Sezer O.، Schmid P.، Shweigert M. و همکاران // برگشت سریع سندرم نفروتیک به دلیل آمیلوئیدوز سیستمیک AL اولیه پس از VAD و متعاقب آن شیمی درمانی با دوز بالا با پشتیبانی فروش ساقه اتولوگ. پیوند مغز استخوان. 1999. - 23 (9): 967-969.
- Sezer O.، Neimoller K.، Jakob C. و همکاران // رویکردهای جدید برای درمان آمیلوئیدوز اولیه. نظر متخصص در مورد غرغرها. 2000. - 9 (10): 2343-2350
- Sezer O.، Eucker J.، Jakob C.، Possinger K. // تشخیص و درمان آمیلوئیدوز AL. کلینیک نفرول. 2000.-53 (6): 417-423.
- اسکینر M. "آمیلوئیدوز" درمان فعلی در آلرژی، ایمونولوژی و روماتولوژی. کتاب Mosby-Year. 1996. - 235-240.
- Palladini G.، Anesi E.، Perfetti V. و همکاران. یک رژیم دگزامتازون با دوز بالا برای آمیلوئیدوز اولیه سیستمیک (AL). مجله هماتولوژی بریتانیا. 2001.-113:1044-1046.
E. V. Zakharova
شهر مسکو بیمارستان بالینیآنها را S. P. Botkina
جدول 2. رژیم های درمانی آمیلوئیدوز اولیه
- تجویز خوراکی دوره ای ملفولان (0.15-0.25 میلی گرم بر کیلوگرم وزن بدن در روز) و پردنیزولون (1.5-2.0 میلی گرم بر کیلوگرم در روز) به مدت چهار تا هفت روز هر چهار تا شش هفته به مدت یک سال، تا زمانی که دوز دوره 600 میلی گرم به دست آید.
- تجویز خوراکیملفولان با دوز 4 میلی گرم در روز به مدت سه هفته، سپس، پس از یک استراحت دو هفته ای - 2-4 میلی گرم در روز چهار روز در هفته به طور مداوم، تا زمانی که دوز دوره 600 میلی گرم در ترکیب با پردنیزولون حاصل شود.
- تزریق داخل وریدی دوزهای بالای ملفولان (100-200 میلی گرم بر متر مربع سطح بدن به مدت دو روز) و سپس پیوند سلول های بنیادی اتولوگ
- دگزامتازون IV 40 میلی گرم به مدت چهار روز هر سه هفته به مدت هشت سیکل
- تزریق داخل وریدی دگزامتازون با دوز 40 میلی گرم در روزهای اول، چهارم، 9-12 و 17-20 سیکل 35 روزه، سه تا شش سیکل، و سپس استفاده از a-interferon با دوز 3-6 میلیون واحد سه بار در هفته.
- طرح وینکریستین-دوکسوریبوسین-دگزامتازون (VAD).
