Amyloidoza układowa: diagnostyka, diagnostyka różnicowa, leczenie. Przebieg kliniczny i jego główne przejawy
amyloidoza- choroba charakteryzująca się zaburzeniem metabolicznym, w wyniku którego powstaje nowa substancja dla organizmu (amyloid), która odkłada się w narządach i zaburza ich funkcje.
Amyloid jest złożoną glikoproteiną, w której białka fibrylarne i globularne są ściśle związane z polisacharydami. Włókno amyloidowe składa się z białek polipeptydowych; oprócz białka włóknistego amyloid zawiera również inne białko - tak zwany składnik P, który jest taki sam dla wszystkich form amyloidu. Uważa się, że składnik P jest normalnym białkiem surowicy związanym z włókienkami amyloidowymi.
Amyloidoza może wystąpić jako powikłanie dowolnej choroby lub rozwinąć się jako niezależny proces.
Obecnie, w zależności od etiologii, istnieje kilka postaci amyloidozy, które mają swój własny skład biochemiczny włókienek amyloidowych.
Amyloidoza pierwotna (idiopatyczna) rozwija się bez wyraźnej przyczyny i atakuje różne narządy (serce, nerki, jelita, wątrobę, układ nerwowy). Biochemiczną postacią pierwotnej amyloidozy jest forma AL, prekursorem takiego amyloidu są łańcuchy lekkie Ig i immunoglobulin. Ze względu na budowę amyloidu i charakter uszkodzenia narządów wewnętrznych, amyloidoza w szpiczaku mnogim jest zbliżona do pierwotnej (idiopatycznej) dawki amyloidu, którą obecnie wyodrębnia się do osobnej grupy.
Dziedziczna (genetyczna) skrobiawica objawia się dominującym uszkodzeniem nerek, połączeniem uszkodzenia nerek i układu nerwowego. W naszym kraju dziedziczna amyloidoza jest zwykle związana z chorobą okresową, która jest przenoszona w sposób autosomalny dominujący. W tej chorobie jedyną manifestacją może być amyloidoza. Biochemiczną postacią dziedzicznej amyloidozy jest AF (prekursorem amyloidu jest prealbumina). W przypadku choroby okresowej formą biochemiczną jest AA (prekursorem jest białko SAA).
Amyloidoza nabyta (wtórna) występuje najczęściej i rozwija się w reumatoidalnym zapaleniu stawów, chorobie Bechterewa, gruźlicy, przewlekłym ropieniu - zapaleniu szpiku, rozstrzeni oskrzeli, przewlekłym ropieniu płuca, rzadziej we wrzodziejącym zapaleniu jelita grubego, łuszczycy, limfogranulomatozy, kiły, nowotworach nerek, płuc i innych Biochemiczną postacią amyloidozy wtórnej jest AA (jej surowiczym prekursorem jest białko SAA syntetyzowane przez hepatocyty).
Amyloidoza starcza jest wynikiem mimowolnych zaburzeń metabolizmu białek, występujących w mózgu, trzustce i sercu. Wzór biochemiczny to AS (prekursorem jest prealbumina).
Miejscowa amyloidoza rozwija się bez wyraźnego powodu, jej wzór biochemiczny to AE (prekursor nieznany).
Patogeneza. Dobrze znane są tylko odrębne ogniwa patogenezy (schemat 22). Ze schematu wynika, że pod wpływem mutacji genu, jak również pod wpływem czynników zewnętrznych, odporność zmienia się – zmniejsza się
liczba limfocytów T. Prowadzi to do zmniejszenia ich kontrolującego wpływu na układ B limfocytów. W rezultacie zmniejsza się liczba komórek B niosących normalne immunoglobuliny, a zwiększa się liczba komórek B syntetyzujących prekursory fibryli amyloidowych. Amyloidoblasty w zwiększonej ilości wytwarzają białko fibrylarne, które powoduje syntezę amyloidu w dużych ilościach.
Jednak ze względu na defekt genetyczny amyloidoklastów, który przyczynia się do zmniejszenia ich aktywności enzymatycznej, nie dochodzi do wystarczającej resorpcji amyloidu. W rezultacie dochodzi do zwiększonego odkładania się amyloidu w tkankach i narządach [Mukhin N.A., 1981].
W szpiczaku mnogim amyloidoza rozwija się w wyniku zwiększonej produkcji paraproteiny przez komórki plazmatyczne, która jest wykorzystywana do budowy amyloidu. Skład amyloidu w różnych postaciach amyloidozy jest różny, co determinuje skład białek włókienek amyloidowych.
Przy uszkodzeniu mięśnia sercowego, nerwów obwodowych (obserwowanych głównie w idiopatycznej postaci amyloidozy), amyloid odkłada się wokół włókien kolagenowych tkanki łącznej. W zmianach obserwuje się odkładanie się amyloidu wokół włókien siatkowatych
nerki, jelita, wątroba, nadnercza, trzustka (z amyloidozą dziedziczną i wtórną). Możliwa jest jednak kombinacja złogów amyloidu okołokolagenowego i okołosiatkówkowego, co zapewnia łączne uszkodzenia różnych narządów i układów.
Wraz z odkładaniem się amyloidu w tkankach zmniejsza się liczba funkcjonujących elementów, kardiomiocytów, hepatocytów, włókien nerwowych i kłębuszków nerkowych, co w konsekwencji prowadzi do rozwoju niewydolności narządowej.
Tak więc w sercu amyloid odkłada się pod wsierdziem, w zrębie i naczyniach mięśnia sercowego, a także wzdłuż żył w nasierdziu. W tym samym czasie serce gwałtownie się powiększa, a liczba kardiomiocytów gwałtownie spada. Wszystko to prowadzi do zmniejszenia funkcji skurczowej mięśnia sercowego i niewydolności serca, a także zaburzeń przewodzenia i rytmu serca. W mózgu z amyloidozą starczą amyloid znajduje się w tzw. blaszkach starczych kory mózgowej, naczyń i błon. W skórze amyloid odkłada się w brodawkach i ścianach naczyń, co prowadzi do gwałtownego zaniku naskórka. W wątrobie amyloid odkłada się między gwiaździstymi retikuloendoteliocytami naczyń sinusoidalnych, w ścianach naczyń, przewodach iw tkance łącznej dróg wrotnych. W miarę gromadzenia się amyloidu komórki wątroby zanikają.
W nerkach amyloid odkłada się w błonie naczyń włosowatych kłębuszków nerkowych i kanalikach nefronu, w mezangium, pętlach naczyń włosowatych i wzdłuż tętniczek. W miarę gromadzenia się amyloidu większość nefronów zanika, obumiera lub jest zastępowana przez tkankę łączną – pojawia się skurczona nerka. Proces ten można przedstawić w postaci następującego diagramu:
białkomocz -> zespół nerczycowy -> niewydolność nerek.
W związku z tym w obrazie klinicznym wyróżnia się trzy etapy: 1) początkowy (białkomocz); 2) wdrożony (nefrotyczny); 3) terminal (azotemiczny).
obraz kliniczny. Objawy amyloidozy są różnorodne i determinowane są przez: 1) lokalizację amyloidu w określonym narządzie; 2) stopień zaawansowania złogów amyloidowych w narządzie; 3) główna choroba, przeciwko której rozwinął się amyloid (z wtórną postacią amyloidozy).
Trudności w rozpoznaniu mogą wynikać z faktu, że objawy kliniczne choroby będą widoczne dopiero przy pewnej ilości złogów amyloidu. Pod tym względem nieunikniony jest okres „utajony” od momentu złogów amyloidu do pojawienia się objawów upośledzenia funkcjonowania narządu lub układu.
Obraz kliniczny jest szczególnie jasny w przypadku uszkodzenia nerek, najczęstszej lokalizacji złogów amyloidowych.
Na pierwszym etapie poszukiwań diagnostycznych na początkowym etapie praktycznie nie można uzyskać informacji wskazujących na uszkodzenie nerek przez amyloidozę. Skargi pacjentów są związane z chorobą podstawową (z wtórną amyloidozą).
W anamnezie znajdują się informacje o występowaniu danej choroby (gruźlica płuc, zapalenie kości i szpiku, reumatoidalne zapalenie stawów itp.), jej przebiegu i leczeniu. Same te informacje nie pozwalają na rozpoznanie amyloidozy nerek, ale zwracają uwagę lekarza na taką możliwość.
W zaawansowanym stadium amyloidozy pacjenci skarżą się, w związku z rozwojem zespołu nerczycowego, na zmniejszenie ilości oddawanego moczu, obrzęki o różnej częstości występowania i nasileniu, a także na osłabienie, brak apetytu i obniżoną sprawność. Wraz z nimi, w amyloidozie wtórnej, pojawiają się skargi na manifestację choroby podstawowej.
W końcowym stadium dolegliwości spowodowane są rozwijającą się przewlekłą niewydolnością nerek: utrata apetytu, nudności, wymioty (zaburzenia dyspeptyczne), bóle głowy, zaburzenia snu (zaburzenia układu nerwowego), świąd skóry.
Na II etapie poszukiwań diagnostycznych we wczesnym stadium można wykryć jedynie objawy charakterystyczne dla choroby podstawowej (z amyloidozą wtórną).
W zaawansowany etap ujawnić: 1) hipostazy o różnej lokalizacji i wyrazistości; przy znacznym zatrzymaniu płynów w organizmie może pojawić się wysięk opłucnowy, hydropericardium, przejściowe wodobrzusze; 2) nadciśnienie tętnicze (występuje u 12-20% chorych na amyloidozę), rozstrzeń i przerost lewej komory; 3) powiększenie wątroby i śledziony z powodu odkładania się amyloidu w tkankach (wątroba i śledziona są gęste, bezbolesne, z zaostrzoną krawędzią); 4) objawy choroby podstawowej (z amyloidozą wtórną).
W etap końcowy objawy zależą od ciężkości niewydolności nerek: 1) zespół dystroficzny (zmiany na skórze i błonach śluzowych); 2) zespół surowiczo-stawowy (osteoartropatia, wtórna dna moczanowa, suche zapalenie osierdzia, zapalenie opłucnej); 3) nadciśnienie tętnicze.
Na III etapie poszukiwania diagnostycznego amyloidozy uzyskuje się najważniejsze informacje do postawienia rozpoznania, które można pogrupować w następujący sposób: 1) zespół moczowy; 2) naruszenia metabolizmu białek i lipidów; 3) wykrywanie złogów mas amyloidowych.
zespół moczowy: 1) białkomocz – najważniejszy objaw amyloidozy, rozwija się we wszystkich jej postaciach, ale najczęściej w amyloidozie wtórnej. Białkomocz jest zwykle znaczny, dziennie uwalniane jest 2-20 g białka, którego główną częścią jest albumina. W mniejszych ilościach globuliny są wydalane, możliwe jest wydalanie z moczem prekursora amyloidu surowicy (białka SAA). W stadium terminalnym białkomocz utrzymuje się. W moczu można wykryć a-, a zwłaszcza y-glikoproteiny.
W zależności od stopnia białkomoczu stwierdza się szkliste i rzadziej ziarniste odlewy. Rzadko rozpoznaje się mikrohematurię lub leukocyturię, ale jej nasilenie nie odpowiada stopniowi białkomoczu (jak obserwuje się przy kłębuszkowym zapaleniu nerek). Stopień zaburzeń metabolizmu lipidów w amyloidozie odpowiada lipoidurii z obecnością dwójłomnych kryształów w osadzie moczu.
Zaburzenia metabolizmu białek i lipidów: 1) hipoproteinemia w połączeniu z hipoalbuminemią i hiper-<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
Ważne dla diagnozy jest wykrywanie mas amyloidowych w narządach i tkankach: w wątrobie (50% przypadków), śledzionie (z biopsją punkcyjną), błonie śluzowej dziąseł i odbytnicy.
W początkowej fazie (białkomocz) biopsja błony śluzowej dziąseł często daje wynik negatywny, a odbytnicy - wynik pozytywny; w zaawansowanym stadium (nerczycowym) w pierwszym przypadku wyniki są dodatnie u połowy pacjentów, aw drugim nawet częściej.
Wreszcie, w przewlekłej niewydolności nerek, dane z biopsji tkanki dziąseł są pozytywne w ponad połowie przypadków, a błony śluzowej odbytnicy w prawie wszystkich przypadkach. Dlatego biopsja błony śluzowej dziąseł powinna być zalecana przy zaawansowanym procesie, a biopsja odbytnicy – w każdym stadium amyloidozy.
Przy podejrzeniu amyloidozy idiopatycznej (występuje częściej przy uszkodzeniu serca, nerwów obwodowych, rzadziej nerek) wskazane jest przede wszystkim wykonanie biopsji błony śluzowej dziąseł, a w przypadku amyloidozy wtórnej (nabytej) i jej formy dziedziczne (występują z dominującym uszkodzeniem nerek) - biopsja błony śluzowej odbytnicy.
Szereg innych badań pomaga: 1) wyjaśnić diagnozę choroby, przeciwko której rozwinęła się amyloidoza; 2) ocenić stan czynnościowy nerek (test Reberga, test Zimnickiego, poziom kreatyniny we krwi).
Pływ. Obraz kliniczny amyloidozy nerkowej ma cechy odróżniające ją od uszkodzenia nerek innego pochodzenia: 1) zespół nerczycowy rozwija się stopniowo i często po długim okresie białkomoczu, charakteryzuje się trwałym przebiegiem, obrzęk często jest oporny na różne leki moczopędne. W przypadku CGN zespół nerczycowy występuje z reguły już na początku choroby i często nawraca w przyszłości; 2) nadciśnienie tętnicze obserwuje się rzadko, nawet w stadium przewlekłej niewydolności nerek; 3) w amyloidozie pierwotnej przewlekła niewydolność nerek przebiega łagodniej, w przeciwieństwie do amyloidozy wtórnej lub CGN (ze względu na mniejsze nasilenie uszkodzenia kłębuszków nerkowych w porównaniu z postaciami wtórnymi amyloidozy); 4) przebieg amyloidozy wtórnej w dużej mierze zależy od choroby podstawowej, z częstymi zaostrzeniami, w których możliwa jest znacząca progresja amyloidozy.
Komplikacje. Z amyloidozą w 2-5% przypadków rozwija się:
1) zakrzepica żył nerkowych (z wtórną amyloidozą), która się objawia
krwiomocz i ból w okolicy lędźwiowej, wzrost białka
ria i spadek diurezy;
2) współistniejąca infekcja;
3) włóknikowo-ropne zapalenie otrzewnej, któremu towarzyszy pojawienie się
następuje gwałtowny wzrost wodobrzusza.
Diagnostyka. Objawy kliniczne amyloidozy są niespecyficzne. Każdy z objawów (obrzęk, białkomocz, nadciśnienie tętnicze) może wystąpić w różnych chorobach nerek. Jedyną metodą wiarygodnego rozpoznania amyloidozy jest biopsja narządu (nerki, wątroby, błony śluzowej odbytu lub dziąseł), jednak nie zawsze jest ona wykonalna. Dlatego w większości przypadków konieczne jest skupienie się na klinicznych objawach procesu patologicznego.
Obecność choroby, w której jest wtórna
amyloidoza (objawy kliniczne lub anamnestyczne).
Początek i postęp białkomoczu lub początek
zespół nerczycowy.
Nie ma choroby, w której może rozwinąć się amyloidoza,
występuje jednak białkomocz lub zespół nerczycowy.
Obecność uporczywej ciężkiej niewydolności serca, zespół nie jest
wystarczająca absorpcja, polineuropatia (jeśli jednocześnie ostatnia
Trudno wytłumaczyć te trzy syndromy innymi przyczynami).
Obecność amyloidozy można podejrzewać za pomocą następujących laboratoryjnych objawów zespołu nerczycowego (który, jak wiadomo, może rozwinąć się wraz z innymi chorobami nerek):
a) ciężka dysproteinemia + hipoalbuminemia + hiper-sg- i gi-
pergammaglobulinemia;
b) wzrost poziomu ag-glikoproteiny, p-lipoproteiny;
c) pojawienie się w moczu a-, a zwłaszcza y-glikoprotein i a-lipopro-
teidy.
We wszystkich przypadkach prawdopodobieństwo rozwoju amyloidozy wzrasta wraz z wykryciem powiększenia wątroby i splenomegalii, a także zmian w sercu charakterystycznych dla amyloidozy (w takich przypadkach mówimy o idiopatycznej uogólnionej amyloidozie).
W związku z tym rozpoznanie amyloidozy nerkowej można postawić z wystarczającą pewnością w stadium zaawansowanym (nerczycowym) lub terminalnym, podczas gdy w stadium początkowym (białkomoczowym) jest to znacznie trudniejsze. W takich przypadkach przemijający lub stały białkomocz należy różnicować z kłębuszkowym zapaleniem nerek (ostrym, przewlekłym). Należy wziąć pod uwagę:
1) wolniejszy postęp uszkodzenia nerek w amyloidzie
dawka;
2) brak wyraźnego związku z przeziębieniem w amyloidozie
niami;
3) stała obecność mikrohematurii w kłębuszkowym zapaleniu nerek (z
amyloidoza w 20% przypadków).
Czasami prawidłowe rozpoznanie można postawić dopiero po okresie długiej obserwacji pacjenta. Problem rozwiązuje się znacznie szybciej, jeśli możliwe jest wykonanie biopsji punkcyjnej nerki.
Formułowanie szczegółowej diagnozy klinicznej amyloidoza uwzględnia następujące elementy: 1) postać amyloidozy; 2) stadium amyloidozy (białkomoczowe, nerczycowe, terminalne); 3) stan czynnościowy nerek (brak lub obecność niewydolności nerek, stopień jej nasilenia); 4) choroba podstawowa (z amyloidozą wtórną); 5) stan innych narządów (serce, wątroba, układ nerwowy itp.) w amyloidozie idiopatycznej (pierwotnej).
Leczenie. Problem leczenia amyloidozy nadal pozostaje nierozwiązany, ponieważ nie wyjaśniono przyczyn prowadzących do wzmożonej amyloidogenezy i niewydolności jej resorpcji. Niemniej jednak możliwe jest przeprowadzenie szeregu działań terapeutycznych poprawiających stan pacjenta. Obecnie leczenie pacjenta z amyloidozą prowadzone jest z uwzględnieniem: 1) wpływu na chorobę podstawową, przeciw której rozwinęła się amyloidoza
(wtórny); 2) wpływ na mechanizmy patogenezy; 3) wpływ na główne zespoły kliniczne.
Wpływ na chorobę podstawową, przeciwko której się rozwija
wtórna amyloidoza, jest to konieczne ze względu na fakt, że często
rozstępy lub wysoka aktywność procesu patologicznego prowadzą do
progresja amyloidozy.
Wpływ ten jest następujący:
a) w przewlekłych infekcjach (gruźlica, kiła)
długotrwała terapia specyficzna;
b) w przewlekłych niespecyficznych chorobach płuc - kom
plexoterapia z antybiotykami, drenażem oskrzeli i
jeśli to konieczne, i interwencja chirurgiczna (na przykład z przewlekłą
ropień płuca);
c) z chorobami ogólnoustrojowymi tkanki łącznej, na przykład z
reumatoidalne zapalenie stawów, wskazana jest kompleksowa terapia, w tym
wartość preparatów podstawowych (D-penicylamina, sole złota, amino-
leki nolinowe).
Wpływ na mechanizmy patogenezy polega na zmniejszeniu
synteza amyloidu:
a) dzienne spożycie 80-120 g surowej wątroby przez 6-12 miesięcy
prowadzi do zmniejszenia białkomoczu, zmniejszenia wielkości wątroby
ani śledziony;
b) preparaty aminochinolinowe (hingamin lub delagil wg
0,25 - 0,5 g dziennie przez wiele miesięcy, a nawet lat)
yut postęp procesu. Najwyraźniej te środki wpływu
yut na temat syntezy włókienek amyloidowych. Leczenie jest tylko skuteczne
we wczesnych stadiach amyloidozy; w zaawansowanym procesie
(szczegółowy zespół nerczycowy, niewydolność nerek)
ness) wyznaczenie tych leków jest niepraktyczne;
c) z rozwojem amyloidozy z powodu okresowej choroby dot
zalecana jest kolchicyna;
d) w pierwotnej amyloidozie przepisywany jest również melfalan, który hamuje
funkcji niektórych klonów limfocytów, w szczególności syn
typowanie immunoglobulin łańcuchów lekkich zaangażowanych w
tworzenie włókienek amyloidowych (jest to również związane z
amyloidoza, która rozwija się w szpiczaku mnogim).
Wpływ na główne zespoły kliniczne obejmuje
eliminacja obrzęków, nadciśnienia tętniczego, a także środki mające na celu
walka z rozwijającą się niewydolnością nerek:
a) z rozwojem zespołu nerczycowego i ciężkiego obrzęku
konieczna jest wystarczająca zawartość białka w pożywieniu, zmniejszenie
gotowana sól, a także wprowadzenie pełnej krwi lub erytrocytów-
noy masa (szczególnie w obecności niedokrwistości), staranne stosowanie
stosowanie leków moczopędnych;
b) nadciśnienie tętnicze rzadko występuje w amyloidozie,
jednak, gdy osiągnie dużą liczbę, konieczne jest wyznaczenie
stosowanie leków przeciwnadciśnieniowych różnych typów;
c) wraz z rozwojem niewydolności nerek leczenie prowadzi się zgodnie z ogólnie przyjętym planem (ograniczenie białka w pożywieniu, przyjmowanie wystarczającej ilości płynów, korekta metabolizmu mineralnego). W niewydolności nerek spowodowanej amyloidozą możliwe jest zastosowanie hemodializy i przeszczepu nerki.
Prognoza. Trudno jest określić czas trwania okresu białkomoczu, jednak po jego wykryciu zwykle po 3 latach rozwija się obrzęk, na tle którego szybko rozwija się CRF. Wszystko to sprawia, że prognoza jest dość poważna.
Zapobieganie. W amyloidozie idiopatycznej i genetycznej nie są znane podstawowe środki zapobiegawcze. W amyloidozie wtórnej profilaktyka polega na leczeniu chorób prowadzących do rozwoju amyloidozy.
RCHD (Republikańskie Centrum Rozwoju Zdrowia Ministerstwa Zdrowia Republiki Kazachstanu)
Wersja: Protokoły kliniczne Ministerstwa Zdrowia Republiki Kazachstanu - 2016 r
Skrobiawica (E85)
Nefrologia
informacje ogólne
Krótki opis
Zatwierdzony
Komisja Wspólna ds. jakości usług medycznych
Ministerstwo Zdrowia i Rozwoju Społecznego Republiki Kazachstanu
z dnia 13 października 2016 r
Protokół nr 13
amyloidoza- grupa chorób, których cechą charakterystyczną jest odkładanie się w tkankach i narządach włóknistej glikoproteiny - amyloidu.
Korelacja między kodami ICD-10 i ICD-9
| ICD-10 | ICD-9 | ||
| Kod | Nazwa | Kod | Nazwa |
| E85 | amyloidoza |
55.23 99.76 |
Zamknięta [przezskórna] [nakłucie] biopsja nerki. Hemodializa. Plazmafereza terapeutyczna Immunoadsorpcja pozaustrojowa |
| E85.0 | Dziedziczna rodzinna amyloidoza bez neuropatii | ||
| E85.1 | Amyloidoza dziedziczna neuropatyczna | ||
| E85.2 | Dziedziczna amyloidoza, nie określona | ||
| E85.3 | Wtórna amyloidoza układowa | ||
| E85.4 | Ograniczona amyloidoza | ||
| E85.8 | Inne postacie amyloidozy | ||
| E85.9 | Amyloidoza, nie określona | ||
Data sporządzenia/weryfikacji protokołu: 2016 r.
Użytkownicy protokołu: lekarze pierwszego kontaktu, terapeuci, hematolodzy, nefrolodzy.
Skala poziomu dowodów:
| ALE | Wysokiej jakości metaanaliza, systematyczny przegląd RCT lub duże RCT z bardzo niskim prawdopodobieństwem (++) błędu systematycznego, którego wyniki można uogólnić na odpowiednią populację. |
| W | Wysokiej jakości (++) systematyczny przegląd badań kohortowych lub kliniczno-kontrolnych lub Wysokiej jakości (++) badań kohortowych lub kliniczno-kontrolnych o bardzo niskim ryzyku błędu systematycznego lub RCT o niskim (+) ryzyku błędu systematycznego, wyniki które można uogólnić na odpowiednią populację. |
| Z |
Badanie kohortowe lub kliniczno-kontrolne lub kontrolowane bez randomizacji z niskim ryzykiem błędu systematycznego (+). których wyniki można uogólnić na odpowiednią populację lub RCT z bardzo niskim lub niskim ryzykiem błędu systematycznego (++ lub +), których wyników nie można bezpośrednio uogólnić na odpowiednią populację. |
| D | Opis serii przypadków lub badania niekontrolowanego lub opinii eksperta. |
Klasyfikacja
Typy amyloidów i odpowiadające im formy amyloidozy:
|
Białko skrobiowaty |
Białko prekursorowe | Postać kliniczna amyloidozy |
| AA | białko SAA | Amyloidoza wtórna w przewlekłych chorobach zapalnych, w tym chorobach okresowych i zespole Muckle-Wellsa |
| glin | λ, κ-łańcuchy lekkie immunoglobulin | Amyloidoza w dyskrazjach plazmocytowych - idiopatyczna, w szpiczaku mnogim i makroglobulinemii Waldenströma |
| ATTR | transtyretyna | Rodzinne formy polineuropatii, kardiopatycznej i innych amyloidoz, układowa amyloidoza starcza |
| Aβ2M | β2-mikroglobulina | amyloidoza dializacyjna |
| AGel | Gelsolina | Fińska rodzinna polineuropatia amyloidowa |
| AApoAI | Apolipoproteina AI | Polineuropatia amyloidowa (typ III, według van Allena, 1956) |
| Migotanie przedsionków | fibrynogen | nefropatia amyloidowa |
| Aβ | β-białko | Choroba Alzheimera, zespół Downa, dziedziczne krwotoki mózgowe z amyloidozą, Holandia |
| APrP Scr | Białko prionowe | choroba Creutzfeldta-Jakoba, choroba Gerstmanna-Strausslera-Scheinkera |
| AANF | Przedsionkowy czynnik natriuretyczny | Izolowana amyloidoza przedsionkowa |
| AIAPP | Amylina | Izolowana amyloidoza w wysepkach Langerhansa w cukrzycy typu II, insulinoma |
| ACal | prokalcytonina | Na raka rdzeniastego tarczycy |
| ACys | Cystatyna C | Dziedziczne krwotoki mózgowe z amyloidozą, Islandia |
Klasyfikacja kliniczna amyloidozy
amyloidoza pierwotna:
pojawiające się bez wyraźnego powodu;
Związany ze szpiczakiem mnogim
amyloidoza wtórna:
w przewlekłych infekcjach;
w reumatoidalnym zapaleniu stawów i innych chorobach tkanki łącznej;
w chorobach onkologicznych;
amyloidoza rodzinna (dziedziczna):
w przypadku okresowej choroby;
odmiana portugalska i inne formy rodzinnej amyloidozy;
amyloidoza starcza
miejscowa amyloidoza
amyloidoza dziedziczna:
neuropatyczny
z uszkodzeniem kończyn dolnych: portugalski, japoński, szwedzki i inne typy;
ze zmianami chorobowymi kończyn górnych: typy Szwajcaria-Indiana, Niemcy-Maryland;
nefropatyczny:
Choroba okresowa
gorączka i ból brzucha u Szwedów i Sycylijczyków;
połączenie wysypki, głuchoty i uszkodzenia nerek;
Uszkodzenie nerek w połączeniu z nadciśnieniem tętniczym;
kardiomiopatyczny:
Duński - postępująca niewydolność serca;
· meksykańsko-amerykański – zespół chorego węzła zatokowego, zatrzymanie przedsionków;
mieszany:
Fiński - dystrofia rogówki i uszkodzenie nerwów czaszkowych;
udar mózgu.
Etapy kliniczne amyloidozy nerek
| Etap | Objaw kliniczny |
| 1 | Etap przedkliniczny lub utajony (bezobjawowy) - amyloid jest obecny w strefie pośredniej, a obrzęk i ogniska stwardnienia rozwijają się wzdłuż bezpośrednich naczyń piramid. Etap trwa 3-5 lub więcej lat. W tym okresie w amyloidozie reaktywnej dominują objawy kliniczne choroby podstawowej (np. proces ropny w płucach, gruźlica, reumatoidalne zapalenie stawów itp.). |
| 2 | Stadium białkomoczu (albuminurię) - amyloid występuje głównie w mezangium, w pętlach naczyń włosowatych, w piramidach i substancji korowej kłębuszków nerkowych, w naczyniach. Rozwija się stwardnienie i zanik nefronów, przekrwienie i limfostaza. Nerki są powiększone i gęste, mają matowy szaro-różowy kolor. Białkomocz na początku jest umiarkowanie wyrażony, może być nawet przejściowy przez pewien czas, zmniejszać się i narastać, ale potem staje się trwały (stadium przerywanego białkomoczu). Niektórzy badacze wyróżniają w tym stadium dwa okresy: białkomocz selektywny i nieselektywny. Czas trwania etapu wynosi od 10 do 13 lat. |
| 3 | Etap nerczycowy (obrzękowy, obrzękowo-hipotoniczny) - nerczyca amyloidowo-lipidowa - amyloid we wszystkich częściach nefronu. Występuje stwardnienie i amyloidoza rdzenia, ale warstwa korowa bez wyraźnych zmian sklerotycznych. Czas trwania etapu wynosi do 6 lat. Zarówno w fazie białkomoczu, jak i nerczycy nerki są powiększone, gęste (duża nerka łojowa). Klinicznie ten etap objawia się klasycznym zespołem nerczycowym ze wszystkimi jego objawami: z rozwojem masywnego białkomoczu (z utratą białka w moczu powyżej 3-5 gramów na dobę), hipoproteinemią z hipoalbuminemią, hipercholesterolemią, lipidurią z obrzęk do stopnia anasarca. W osadzie moczu znajdują się wałeczki hialinowe, a wraz ze wzrostem białkomoczu pojawiają się wałeczki ziarniste. Możliwe mikro- i makrohematuria, leukocyturia bez objawów odmiedniczkowego zapalenia nerek. |
| 4 | Etap mocznicowy (końcowy, azotemichesky) - nerka pomarszczona amyloidem - zmniejszona, gęsta, bliznowata nerka. Przewlekła niewydolność nerek niewiele różni się od innych chorób nerek. Uważa się, że w przeciwieństwie do kłębuszkowego zapalenia nerek, w którym początek CRF występujący z wielomoczem może prowadzić do przynajmniej częściowej konwergencji obrzęku, w amyloidozie azotemia rozwija się na tle niskiego ciśnienia krwi i zespołu nerczycowego. |
Diagnostyka (przychodnia)
DIAGNOSTYKA NA POZIOMIE Ambulatoryjnym
Kryteria diagnostyczne
Uskarżanie się:
Osłabienie, zwiększone zmęczenie;
· ból głowy;
obrzęk nóg, ramion i twarzy;
wysokie ciśnienie krwi;
nudności, biegunka (biegunka);
ból w okolicy serca;
ból w mięśniach.
Anamneza:
· utrata masy ciała;
Obecność gammopatii monoklonalnej nieznanego pochodzenia;
przewlekłe choroby zapalne (ropne);
przewlekłe infekcje;
dziedziczność.
Badanie lekarskie
Generalna Inspekcja:
Plamica okołooczodołowa (obserwowana w 15% przypadków);
makroglossia jest charakterystyczna dla pierwotnej amyloidozy (AL);
Duszność podczas wysiłku fizycznego (obserwowana u około 40% pacjentów);
objaw barkowy (naciek okołostawowy amyloidu prowadzi do fałszywego przerostu i zwiększenia objętości mięśni obręczy barkowej i uda).
Osłuchiwanie:
możliwa obecność zaburzeń rytmu serca.
Palpacja:
obrzęki kończyn dolnych, spowodowane hipoalbuminemią i zespołem nerczycowym, a także zastojem krążenia ogólnoustrojowego z powodu kardiomiopatii restrykcyjnej (obserwowane w 50% przypadków);
powiększenie wątroby i śledziony;
parestezje (obserwowane u około 15% pacjentów);
Spastyczny ból w przewodzie pokarmowym;
Może wystąpić wzrost ślinianek podżuchwowych.
Badania laboratoryjne:
Pełna morfologia krwi - niedokrwistość, leukocytoza, zwiększona ESR;
ogólna analiza moczu - białkomocz, mikrohematuria, aseptyczna leukocyturia;
Biochemiczne badanie krwi (białko całkowite, albumina, Na, Ca, cholesterol, cukier w surowicy krwi) - hipoproteinemia (z powodu hipoalbuminemii), hiperglobulinemia, hiponatremia, hipoprotrombinemia, hipokalcemia, hipercholesterolemia.
USG narządów jamy brzusznej i nerek - wizualizowane są powiększone, zwarte nerki (duże nerki tłuszczowe).

Diagnostyka (szpital)
DIAGNOSTYKA NA POZIOMIE STACJONARNYM
Kryteria diagnostyczne na poziomie szpitalnym
Skargi i anamnezy: patrz poziom ambulatoryjny .
Badanie lekarskie: patrz poziom ambulatoryjny .
Badania laboratoryjne:
| test diagnostyczny | Wynik |
|
Immunofiksacja surowicy Wynik testu jest dodatni u 60% pacjentów z amyloidozą z immunoglobuliną łańcucha lekkiego (AL) (6). |
|
|
Immunofiksacja moczu Wynik testu jest dodatni u 80% pacjentów z amyloidozą AL (6). Wykrycie w moczu białka łańcucha lekkiego sugeruje obecność szpiczaka mnogiego i amyloidozy. |
Obecność białka monoklonalnego |
|
Test immunoglobulin wolnych od łańcuchów lekkich w surowicy Ten stosunkowo nowy test ma bardzo wysoką czułość (>95%) w diagnostyce amyloidozy AL (10). Dostępne w handlu surowice odpornościowe przeciwko łańcuchom lekkim immunoglobulin, AA i transtyretynie są powszechnie stosowane, ale mogą nie mieć wystarczającej specyficzności i czułości. W wielu przypadkach do określenia podstawowego typu amyloidu potrzebna jest spektroskopia masowa i mikroskopia immunoelektronowa. |
Nieprawidłowy współczynnik kappa lambda |
|
Biopsja szpiku kostnego Biopsja szpiku kostnego jest wykonywana u wszystkich pacjentów z podejrzeniem amyloidozy łańcuchów lekkich i jest doskonałym źródłem tkanki do diagnozowania każdego pacjenta z podejrzeniem amyloidozy. |
Obecność klonalnych komórek plazmatycznych |
Badania instrumentalne:
USG narządów jamy brzusznej i nerek - wizualizowane są powiększone, zwarte nerki (duże nerki tłuszczowe).
Algorytm diagnostyczny amyloidozy nerek
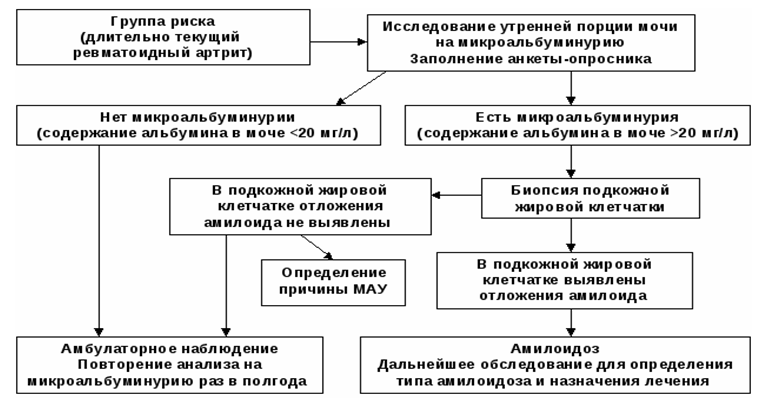
Lista głównych środków diagnostycznych:
· ogólna analiza krwi;
· ogólna analiza moczu;
analiza biochemiczna krwi (białko całkowite, albumina, Na, Ca, cholesterol, cukier w surowicy krwi);
immunofiksacja surowicy;
immunofiksacja moczu;
Badanie wolnych łańcuchów lekkich immunoglobulin w surowicy;
biopsja szpiku kostnego.
USG narządów jamy brzusznej i nerek.
Wykaz dodatkowych środków diagnostycznych
Badania laboratoryjne:
| test diagnostyczny | Wynik |
|
Biopsja tkanki: Do rozpoznania amyloidozy konieczne jest, aby złogi w tkankach w materiale biopsyjnym zabarwiły się pozytywnie na czerwień Kongo (11). Jasnozieloną dwójłomność można zobaczyć, gdy materiał Kongo jest zabarwiony na czerwono w świetle spolaryzowanym. Materiał do biopsji można pobrać z błony śluzowej warg, skóry, dziąseł, tłuszczu podskórnego, szpiku kostnego, nerwów, odbytnicy, nerek, wątroby lub serca. Złogi są zawsze zlokalizowane pozakomórkowo i są amorficzne. |
pozytywna - zielona dwójłomność przy barwieniu czerwienią Kongo |
|
Badania immunohistologiczne złogów amyloidowych: Pozwalają rozpoznać różne formy amyloidozy układowej. |
surowica odpornościowa przeciwko immunoglobulinie łańcucha lekkiego, AA i transtyretynie |
|
Masa - spektroskopia: Zapewnia analizę składu białek amyloidowych. Jest to obecnie złoty standard w diagnostyce typu amyloidozy. |
potwierdza rodzaj białka |
|
Mikroskopia immunoelektronowa: Wszystkie formy amyloidu pod mikroskopem elektronowym są włókniste, sztywne i nierozgałęzione. |
amyloidy mają wygląd włóknisty, są sztywne i nierozgałęzione. |
|
Badania genetyczne: Testy genetyczne są obowiązkowe w celu wykluczenia dziedzicznej amyloidozy w przypadku wątpliwych wyników testu wykrywania białek monoklonalnych immunoglobulin/amyloidów wolnych łańcuchów lekkich. Geny można badać przez bezpośrednie sekwencjonowanie i obejmują one następujące geny: TTR, fibrynogen, apolipoproteina A1, lizozym, MEFV (gorączka śródziemnomorska) i receptor czynnika martwicy nowotworu 1 (TNFR1 lub TNFRSF1A). MEFV i TNFRSF1A są dziedzicznymi zespołami okresowej gorączki (tj. potencjalnymi przyczynami amyloidozy AA) i nie są dziedzicznymi amyloidozami per se. |
pozytywny |
|
Skan scyntygraficzny surowicy amyloidu P (SAP): W ostatnich latach w praktyce klinicznej zaczęto stosować scyntygrafię składnika P (SAP) znakowaną jodem do oceny dystrybucji amyloidu w organizmie. |
wychwyt w miejscach odkładania się amyloidu |
|
Ogólna analiza krwi: Niedokrwistość występuje głównie u pacjentów z niewydolnością nerek lub krwawieniem z przewodu pokarmowego. Nadpłytkowość jest związana z zajęciem wątroby i hipersplenizmem. |
zwykle normalne |
|
Biochemiczne badanie krwi (badania wątroby i nerek), wskaźniki stanu metabolicznego): Amyloidoza wątrobowa charakteryzuje się podwyższonym poziomem fosfatazy alkalicznej. Większość pacjentów we wczesnych stadiach amyloidozy nerek zachowuje klirens kreatyniny, ale może wystąpić znaczny stopień hipoalbuminemii z powodu utraty białka w moczu (zespół nerczycowy). |
Niska albumina; wzrost fosfatazy alkalicznej |
|
Białkomocz dobowy (pobranie moczu w ciągu 24 godzin): Wydalanie albumin > 1 g/dobę u pacjentów z amyloidozą wskazuje na uszkodzenie nerek (amyloidoza nerek). Przy poziomie białkomoczu > 3 g/dobę rozwija się zespół nerczycowy. |
zwiększone stężenie białka w moczu |
|
Poziom troponiny w surowicy: Czuły test do określania uszkodzenia mięśnia sercowego. Pacjenci z wykrywalnym stężeniem troponiny mają gorsze rokowanie niż ci bez niego (12). |
podniesiony |
|
Peptyd natriuretyczny typu B: Czułe badanie diagnostyczne na obecność rozdęcia mięśnia sercowego i CHF. Wykazano, że ma ważną wartość prognostyczną w ustalaniu amyloidozy serca (13). Na poziomie peptydu natriuretycznego typu B > 300 ng/L (> 300 pg/ml) sugeruje zajęcie amyloidu mięśnia sercowego (10). Pacjenci z<170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с >170 ng/l (> 170 pg/ml). |
podniesiony |
|
mikroglobuliny beta-2: Jest predyktorem przeżycia u pacjentów z amyloidozą. Przy poziomie beta-2-mikroglobuliny > 2,7 mg/l rokowanie jest niekorzystne (14). |
podniesiony |
Badania instrumentalne:
|
EKG: Należy wykonać u wszystkich pacjentów w ramach oceny zajęcia serca. |
zaburzenia przewodzenia serca |
|
Echokardiogram (EchoCG): Objawy kliniczne niewydolności serca u pacjentów z amyloidozą serca obserwuje się od 22% do 34% (7). Echokardiografia ujawnia dużą częstość odkładania się amyloidu u pacjentów z minimalnymi objawami (około 50% przypadków AL dotyczy serca). W ostatnim etapie następuje spadek frakcji wyrzutowej. |
dysfunkcja rozkurczowa, pogrubienie przegrody międzykomorowej, zmniejszenie frakcji wyrzutowej |
|
Echo Dopplera z napięciem: Wskaźnik stopnia nacieku amyloidu w mięśniu sercowym. Posiada wysoką czułość w wykrywaniu nieprawidłowości przy braku nadciśnienia tętniczego lub wad zastawkowych serca. Rozciągnięcie mięśnia sercowego definiuje się jako procentową zmianę długości włókien mięśnia sercowego na jednostkę długości, a szybkość zależy od czasu trwania rozciągnięcia (15–16). |
Zmniejszenie podłużnego skurczu i rozciągnięcia mięśnia sercowego; ograniczenie napełniania komór |
|
MRI serca: Relaksometria rezonansu magnetycznego poprawia wiarygodność diagnostyki MRI i pomaga odróżnić amyloidozę serca od kardiomiopatii przerostowej. |
znacząco wydłużyły czasy relaksacji T1 i T2 w porównaniu z grupą kontrolną |
Diagnostyka różnicowa
Diagnostyka różnicowa amyloidozy nerek
| Państwo | Znaki/symptomy różniczkowe | Testy różniczkowalne |
| Kardiomiopatia przerostowa | Klinicznie trudno jest odróżnić HCM od amyloidozy serca. |
EchoCG jest kryterium diagnostycznym HCM, w którym stwierdza się asymetryczny przerost przegrody międzykomorowej; Echo dopplerowskie z napięciem, stosowane w celu wykluczenia cech amyloidozy, nie wskazuje na typowe restrykcyjne zmiany wypełnienia wykrywane w amyloidozie; MRI może rozróżnić 2 zespoły |
| Błoniasta glomerulopatia | Klinicznie identyczne objawy u pacjentów z zespołem nerczycowym. | Biopsja nerki nie barwi się czerwienią Kongo. |
| Neuropatia związana z gammapatią monoklonalną nieznanego pochodzenia (MGNG). | Pacjenci nie mają znacznego stopnia białkomoczu, hepatomegalii ani kardiomiopatii. | biopsja nerwu łydkowego nie barwi się czerwienią Kongo. |
| szpiczak mnogi | Ból kości, objawy niedokrwistości i niewydolności nerek. |
Zwykłe zdjęcia rentgenowskie pokazują lityczne zmiany kostne, złamania kompresyjne, rozlaną osteoporozę; niska Hb; niewydolność nerek. |
| zespół nerczycowy | Białkomocz dzienny powyżej 3,5 g/dobę, obrzęki, hipoalbuminemia, dyslipidemia | patrz KP „Zespół nerczycowy” |
Turystyka medyczna
Uzyskaj leczenie w Korei, Izraelu, Niemczech, USA
Turystyka medyczna
Uzyskaj porady dotyczące turystyki medycznej
Leczenie
Uwaga!
- Samoleczenie może spowodować nieodwracalne szkody dla zdrowia.
- Informacje zamieszczone na stronie MedElement nie mogą i nie powinny zastępować osobistej konsultacji lekarskiej. Koniecznie skontaktuj się z placówkami medycznymi, jeśli masz jakiekolwiek choroby lub objawy, które Cię niepokoją.
- Wybór leków i ich dawkowanie należy omówić ze specjalistą. Tylko lekarz może przepisać odpowiedni lek i jego dawkowanie, biorąc pod uwagę chorobę i stan organizmu pacjenta.
- Witryna MedElement jest wyłącznie źródłem informacji i materiałów referencyjnych. Informacje zamieszczone na tej stronie nie powinny być wykorzystywane do samowolnej zmiany zaleceń lekarskich.
- Redakcja MedElement nie ponosi odpowiedzialności za uszczerbek na zdrowiu lub szkody materialne wynikające z korzystania z tej strony.
- ogólna, ogólnoustrojowa choroba organizmu, w której dochodzi do odkładania się określonej glikoproteiny (amyloidu) w narządach i tkankach z upośledzoną funkcją tych ostatnich. Amyloidoza może wpływać na nerki (zespół nerczycowy, zespół obrzękowy), serce (niewydolność serca, arytmie), przewód pokarmowy, układ mięśniowo-szkieletowy i skórę. Być może rozwój zapalenia błon surowiczych, zespołu krwotocznego, zaburzeń psychicznych. Wiarygodne rozpoznanie amyloidozy ułatwia wykrycie amyloidu w próbkach biopsyjnych dotkniętych tkanek. W leczeniu amyloidozy przeprowadza się terapię immunosupresyjną i objawową; według wskazań - dializy otrzewnowe, przeszczepy nerek i wątroby.
ICD-10
E85

Informacje ogólne
Amyloidoza jest chorobą z grupy dysproteinoz układowych, która występuje z powstawaniem i gromadzeniem się w tkankach złożonego związku białkowo-polisacharydowego - amyloidu. Częstość występowania amyloidozy na świecie jest w dużej mierze uwarunkowana geograficznie: na przykład choroby okresowe częściej występują w krajach basenu Morza Śródziemnego; polineuropatia amyloidowa - w Japonii, Włoszech, Szwecji, Portugalii itp. Średnia częstość występowania amyloidozy w populacji wynosi 1 przypadek na 50 tysięcy ludności. Choroba zwykle rozwija się u osób w wieku powyżej 50-60 lat. Biorąc pod uwagę fakt, że amyloidoza dotyczy prawie wszystkich układów narządów, choroba jest badana przez różne dyscypliny medyczne: reumatologię, urologię, kardiologię, gastroenterologię, neurologię itp.
Przyczyny amyloidozy
Etiologia pierwotnej amyloidozy nie jest w pełni poznana. Jednocześnie wiadomo, że wtórna amyloidoza jest zwykle związana z przewlekłymi chorobami zakaźnymi (gruźlica, kiła, promienica) i ropnymi chorobami zapalnymi (zapalenie kości i szpiku, rozstrzenie oskrzeli, bakteryjne zapalenie wsierdzia itp.), Rzadziej z procesami nowotworowymi (limfogranulomatoza, białaczka , rak trzewny). narządy). Amyloidoza reaktywna może rozwinąć się u pacjentów z miażdżycą, łuszczycą, chorobami reumatologicznymi (reumatoidalne zapalenie stawów, zesztywniające zapalenie stawów kręgosłupa), przewlekłymi stanami zapalnymi (wrzodziejące zapalenie jelita grubego, choroba Leśniowskiego-Crohna), zmianami wieloukładowymi (choroba Whipple'a, sarkoidoza). Wśród czynników przyczyniających się do rozwoju amyloidozy pierwszorzędne znaczenie ma hiperglobulinemia, upośledzenie funkcjonowania odporności komórkowej, predyspozycje genetyczne itp.
Patogeneza
Wśród licznych wersji amyloidogenezy najwięcej zwolenników ma teoria dysproteinozy, lokalna geneza komórkowa, teorie immunologiczne i mutacyjne. Teoria lokalnej genezy komórkowej uwzględnia jedynie procesy zachodzące na poziomie komórkowym (tworzenie włóknistych prekursorów amyloidu przez układ makrofagów), podczas gdy powstawanie i akumulacja amyloidu zachodzi poza komórką. Dlatego teorii lokalnej genezy komórkowej nie można uznać za wyczerpującą.
Zgodnie z teorią dysproteinozy amyloid jest produktem nieprawidłowego metabolizmu białek. Główne ogniwa w patogenezie amyloidozy - dysproteinemia i hiperfibrynogenemia przyczyniają się do gromadzenia się frakcji gruboziarnistych białek i paraprotein w osoczu. Immunologiczna teoria pochodzenia amyloidozy wiąże powstawanie amyloidu z reakcją antygen-przeciwciało, w której rolę antygenów pełnią obce białka lub produkty rozpadu własnych tkanek. W tym przypadku odkładanie się amyloidu następuje głównie w miejscach powstawania przeciwciał i nadmiaru antygenów. Najbardziej uniwersalna jest teoria mutacji amyloidozy, która uwzględnia ogromną różnorodność czynników mutagennych, które mogą powodować nieprawidłową syntezę białek.
Amyloid jest złożoną glikoproteiną składającą się z białek fibrylarnych i kulistych ściśle związanych z polisacharydami. Złogi amyloidowe gromadzą się w błonie wewnętrznej i przydance naczyń krwionośnych, zrębie narządów miąższowych, strukturach gruczołowych itp. Przy niewielkich złogach amyloidowych zmiany wykrywane są jedynie na poziomie mikroskopowym i nie prowadzą do zaburzeń czynnościowych. Wyraźnemu nagromadzeniu amyloidu towarzyszą zmiany makroskopowe w zajętym narządzie (zwiększenie objętości, tłusty lub woskowaty wygląd). W wyniku amyloidozy, stwardnienia podścieliska i zaniku miąższu narządów dochodzi do ich istotnego klinicznie upośledzenia czynnościowego.
Klasyfikacja
Zgodnie z przyczynami wyróżnia się amyloidozę pierwotną (idiopatyczną), wtórną (reaktywną, nabytą), dziedziczną (rodzinną, genetyczną) i starczą. Wyróżnia się różne formy amyloidozy dziedzicznej: gorączkę śródziemnomorską, czyli choroby okresowe (napady gorąca, bóle brzucha, zaparcia, biegunki, zapalenie opłucnej, zapalenie stawów, wysypki skórne), amyloidozę neuropatyczną portugalską (polineuropatia obwodowa, impotencja, zaburzenia przewodzenia w sercu), typ fiński ( zanik rogówki, neuropatia czaszkowa), wariant duński (kardiopatyczna skrobiawica) i wiele innych. inni
W zależności od dominujących uszkodzeń narządów i układów, nefropatyczne (amyloidoza nerek), kardiopatyczne (amyloidoza serca), neuropatyczne (amyloidoza układu nerwowego), hepatopatyczne (amyloidoza wątroby), epinefropatyczne (amyloidoza nadnerczy) ), wyróżnia się amyloidozę APUD, amyloidozę skóry i mieszany typ choroby. . Ponadto w praktyce międzynarodowej zwyczajowo rozróżnia się amyloidozę miejscową i uogólnioną (układową). Zlokalizowane formy, zwykle rozwijające się u osób starszych, obejmują amyloidozę w chorobie Alzheimera, cukrzycę typu 2, guzy endokrynologiczne, nowotwory skóry, pęcherza moczowego itp. W zależności od składu biochemicznego włókienek amyloidowych wyróżnia się następujące układowe formy amyloidozy :
- glin- jako część fibryli łańcuchów lekkich Ig (przy chorobie Waldenströma, szpiczaku mnogim, chłoniakach złośliwych);
- AA- jako część fibryli, α-globulina surowicy ostrej fazy, podobna pod względem właściwości do białka C-reaktywnego (w przypadku chorób nowotworowych i reumatycznych, chorób okresowych itp.);
- Aβ2M- jako część fibryli β2-mikroglobuliny (w przewlekłej niewydolności nerek u pacjentów poddawanych hemodializie);
- ATTR- w składzie fibryli białko transportowe transtyretyna (w rodzinnych dziedzicznych i starczych postaciach amyloidozy).
Objawy amyloidozy
Objawy kliniczne amyloidozy są zróżnicowane i zależą od nasilenia i lokalizacji złogów amyloidowych, składu biochemicznego amyloidu, „doświadczenia” choroby oraz stopnia dysfunkcji narządowej. W utajonym stadium amyloidozy, gdy złogi amyloidu można wykryć jedynie mikroskopowo, nie występują żadne objawy. Wraz z rozwojem i postępem niewydolności czynnościowej jednego lub drugiego narządu, objawy kliniczne choroby nasilają się.
W przypadku amyloidozy nerek długotrwały obecny etap umiarkowanego białkomoczu zostaje zastąpiony rozwojem zespołu nerczycowego. Przejście do zaawansowanego stadium może wiązać się ze współistniejącą infekcją, szczepieniem, hipotermią, zaostrzeniem choroby podstawowej. Stopniowo narastają obrzęki (najpierw na nogach, a później na całym ciele), rozwija się nefrogenne nadciśnienie tętnicze i niewydolność nerek. Może wystąpić zakrzepica żył nerkowych. Masywnej utracie białka towarzyszy hipoproteinemia, hiperfibrynogenemia, hiperlipidemia i azotemia. W moczu stwierdza się mikro-, czasem makrohematurię, leukocyturię. Ogólnie, podczas amyloidozy nerek wyróżnia się wczesny etap bezobrzękowy, etap obrzękowy i etap mocznicowy (kachektyczny).
Amyloidoza serca przebiega zgodnie z rodzajem kardiomiopatii restrykcyjnej z typowymi objawami klinicznymi - kardiomegalią, arytmią, postępującą niewydolnością serca. Pacjenci skarżą się na duszność, obrzęk, osłabienie, które występuje przy niewielkim wysiłku fizycznym. Rzadziej z amyloidozą serca rozwija się zapalenie błon surowiczych (wodobrzusze, wysiękowe zapalenie opłucnej i zapalenie osierdzia).
Klęska przewodu pokarmowego w amyloidozie charakteryzuje się naciekiem amyloidu języka (macroglassia), przełyku (sztywność i zaburzenia perystaltyki), żołądka (zgaga, nudności), jelit (zaparcia, biegunka, zespół złego wchłaniania, niedrożność jelit). Krwawienie z przewodu pokarmowego może wystąpić na różnych poziomach. Wraz z naciekiem amyloidu w wątrobie rozwija się hepatomegalia, cholestaza i nadciśnienie wrotne. Zajęcie trzustki w amyloidozie zwykle przybiera postać przewlekłego zapalenia trzustki.
Amyloidoza skóry występuje z pojawieniem się wielu blaszek woskowych (grudki, guzki) na twarzy, szyi, naturalnych fałdach skórnych. Zewnętrznie zmiany skórne mogą przypominać twardzinę skóry, neurodermit lub liszaj płaski. W przypadku zmian amyloidowych układu mięśniowo-szkieletowego typowy jest rozwój symetrycznego zapalenia wielostawowego, zespołu cieśni nadgarstka, zapalenia okołostawowego kości ramienno-łopatkowej i miopatii. Oddzielnym postaciom amyloidozy, występującym z zajęciem układu nerwowego, może towarzyszyć polineuropatia, porażenie kończyn dolnych, bóle głowy, zawroty głowy, niedociśnienie ortostatyczne, potliwość, otępienie itp.
Diagnostyka
Z klinicznymi objawami amyloidozy mogą zetknąć się różni klinicyści: reumatolodzy, urolodzy, kardiolodzy, gastroenterolodzy, neurolodzy, dermatolodzy, terapeuci itp. Kompleksowa ocena objawów klinicznych i anamnestycznych, kompleksowe badanie laboratoryjne i instrumentalne mają ogromne znaczenie dla prawidłowego rozpoznania.
Amyloidoza jest chorobą ogólnoustrojową, gdy zaburzony jest metabolizm białek, układ odpornościowy przestaje działać. W związku z tym powstaje amyloid - kompleks białkowo-sacharydowy, który osadza się we wszystkich tkankach narządów ludzkich.
Z biegiem czasu amyloid coraz bardziej atakuje narządy, wypierając normalne komórki. W efekcie narząd traci swoją funkcjonalność, obserwuje się nieodwracalne zmiany. Jeśli choroba nie jest leczona przez długi czas, funkcje kilku narządów są zaburzone, co prowadzi do śmierci.
Według badań WHO amyloidozę rozpoznaje się u około 1% mieszkańców świata. Najczęstszą jest amyloidoza wtórna. Amyloidozę genetyczną częściej diagnozuje się u osób narodowości żydowskiej, ormiańskiej, a także u mieszkańców krajów basenu Morza Śródziemnego.
Wskaźnik zachorowalności wśród mężczyzn jest dwukrotnie wyższy niż u kobiet. Spośród wszystkich postaci amyloidozy rozpoznaje się amyloidozę nefropatyczną (uszkodzenie nerek) i uogólnioną (uszkodzenie wszystkich tkanek i narządów).
Rodzaje amyloidozy, przyczyny rozwoju
W zależności od przyczyny amyloidozy istnieją rodzaje chorób, które mogą rozwijać się niezależnie lub z powodu patologii w innych układach i narządach. Występują następujące typy skrobiawicy: starcza, z guzami, amyloidoza pierwotna lub idiopatyczna, dziedziczna, wtórna lub reaktywna, a także u pacjentów poddawanych hemodializie. W zależności od gatunku rozwój amyloidozy przebiega różnie, różnią się objawy i rokowanie. Rodzaje i etapy amyloidozy zostaną szczegółowo omówione poniżej.
Pierwotne (idiopatyczne)
Pierwotna amyloidoza w większości przypadków zaczyna się bez powodu. W tej postaci choroby amyloid odkłada się w tkankach i narządach, obserwuje się mutację komórek układu odpornościowego. Powstały w tym procesie AL-amyloid gromadzi się w mięśniach, skórze, układzie sercowo-naczyniowym i nerwach. Również AL-amyloid powstaje na tle szpiczaka nowotworowego, gdy komórki plazmatyczne zaczynają wydzielać globuliny w dużej objętości. Po związaniu się z nukleoproteinami osocza nieprawidłowe globuliny są przekształcane w amyloid.
Wtórny (reaktywny)
Wtórna amyloidoza rozwija się na tle postępujących procesów zapalnych w czasie. W tym przypadku powstaje AA-amyloid jako powikłanie innych chorób. Przyczynami wtórnej amyloidozy są:
- Przewlekłe infekcje - trąd, malaria, gruźlica, kiła, odmiedniczkowe zapalenie nerek, rozstrzenie oskrzeli.
- Ropne choroby przewlekłe - ropienie ran przez długi czas, zapalenie kości i szpiku.
- Nowotwory - białaczka, limfogranulomatoza itp.
- Obecność niespecyficznego wrzodziejącego zapalenia jelita grubego (zapalenie jelita grubego).
- Choroby reumatologiczne - zesztywniające zapalenie stawów kręgosłupa, reumatoidalne zapalenie stawów itp.
Amyloidoza wtórna może wpływać na każdy narząd, tkankę w ciele. Manifestacja obrazu choroby nie jest od razu zauważalna. Po latach od wystąpienia choroby podstawowej można zauważyć naruszenie funkcji narządu, w którym przede wszystkim odkładał się amyloid. Częściej choroba ta dotyczy wątroby, nerek, śledziony i węzłów chłonnych. Z biegiem czasu zajęte są również inne narządy, co prowadzi do niewydolności wielonarządowej i śmierci.
dziedziczna amyloidoza
Dziedziczna postać amyloidozy wynika z obecności zmutowanych genów w komórkach układu odpornościowego. Te mutacje genetyczne są przekazywane z pokolenia na pokolenie, w wyniku czego powstają amyloidoblasty. Forma dziedziczna dotyka ludzi na określonym obszarze lub należących do określonej grupy etnicznej. Dziedziczna amyloidoza dzieli się na typy:
- Kardiopatyczny. Najczęściej diagnozowana u mieszkańców Danii. Obraz kliniczny choroby przypomina pierwotną amyloidozę typu uogólnionego.
- neuropatyczny. Charakteryzuje się uszkodzeniem tkanki nerwowej. W zależności od lokalizacji zmiany wyróżnia się amyloidozę portugalską (nerwy nóg), amerykańską (nerwy rąk), fińską (układ nerwowy, rogówki oczu, nerki).
- Rodzinna nefropatia. Inna nazwa to amyloidoza angielska (choroba Muckle'a i Wellsa). Obraz kliniczny to pokrzywka, napady gorączki, utrata słuchu.
- Okresowo (rodzinna gorączka śródziemnomorska). Choroba częściej występuje wśród Żydów, Arabów, Ormian. Manifestacje - temperatura powyżej 39ºС, ból głowy i mięśni, obfite pocenie się. Występuje zapalenie błon płucnych, narządów otrzewnej, błony maziowej. Częste odchylenia w psychice.
Amyloidoza starcza
U osób, które ukończyły 80 lat, amyloid odkłada się miejscowo w różnych tkankach i narządach. Choroba jest powiązana z innymi chorobami związanymi z wiekiem. Istnieją dwa rodzaje amyloidozy starczej:
- Mózgowy lub mózgowy. Rozwija się na tle choroby Alzheimera. Amyloid Ab odkłada się w tkankach mózgowych.
- Serdeczny. Może wpływać na komory serca (gdy amyloid powstaje ze zmutowanej transtyretyny białka krwi) i przedsionki (gdy amyloid powstaje z peptydu natriuretycznego wydzielanego przez komórki serca). W obu przypadkach amyloidy znajdują się w tkankach płuc, trzustki i śledziony.
Na nowotwory
Niektóre rodzaje nowotworów wpływają na transformację złośliwą komórek chorego narządu, które w efekcie wytwarzają białko fibrylarne. W tym przypadku amyloidoza rozwija się lokalnie w tkance narządu dotkniętego nowotworem. Przyczyny wywołujące amyloidozę w nowotworach:
- Guz rdzeniasty tarczycy. Rak rozwija się z komórek C tarczycy, które normalnie są odpowiedzialne za produkcję kalcytocyny. Kiedy synteza kalcytocyny jest zaburzona, jej fragmenty stają się częścią amyloidu AE.
- Rak wysepek tarczycy. Wysepki to skupiska komórek odpowiedzialnych za produkcję hormonów – glukagonu, insuliny, somatostatyny itp. Złośliwe zwyrodnienie komórek powoduje uwolnienie białka włóknistego, które następnie ulega degeneracji do amyloidu.
Amyloidoza w hemodializie
 Hemodializa nazywana jest procedurą ratującą życie dla pacjentów, których nerki nie są w stanie oczyścić krwi z toksyn, ubocznych produktów przemiany materii. Przypisz hemodializę osobom, u których zdiagnozowano niewydolność nerek (ostra, przewlekła).
Hemodializa nazywana jest procedurą ratującą życie dla pacjentów, których nerki nie są w stanie oczyścić krwi z toksyn, ubocznych produktów przemiany materii. Przypisz hemodializę osobom, u których zdiagnozowano niewydolność nerek (ostra, przewlekła).
Istotą zabiegu jest przejście krwi przez aparat, który usuwa z niej szkodliwe substancje, powrót oczyszczonej krwi do organizmu pacjenta.
Podczas dializy mikroglobulina B2 nie może być wydalana z organizmu, a jeśli pacjent jest zmuszany do poddania się hemodializie przez długi czas, białko gromadzi się w organizmie w nadmiernych ilościach. Wiąże się z nukleoproteinami osocza, osiada w różnych narządach i staje się podstawą amyloidu.
Objawy amyloidozy
Biorąc pod uwagę, że choroba może rozprzestrzenić się na dowolny narząd lub tkankę, objawy będą się różnić. Różne postacie choroby na początku przebiegu charakteryzują się uszkodzeniem i dysfunkcją jednego narządu w organizmie człowieka.
Z biegiem czasu choroba (jeśli nie jest to amyloidoza miejscowa) postępuje, atakując inne narządy i tkanki. Objawy amyloidozy można zaobserwować w nerkach, wątrobie, sercu i nadnerczach, śledzionie, przewodzie pokarmowym i układzie nerwowym, stawach, mięśniach i skórze. Rodzaje chorób opisano szczegółowo poniżej.
Uszkodzenie nerek
Amyloidoza nerek jest uważana za najniebezpieczniejszą chorobę w porównaniu z uszkodzeniem innych narządów. Obraz kliniczny amyloidozy nerki zależy od stadium. W sumie jest ich 4 - utajony, nerczycowy, azotemiczny, białkomoczowy.
W stadium utajonym amyloidoza nerek praktycznie nie daje objawów. Jeśli jest to postać wtórna, pacjent odczuwa objawy choroby podstawowej. Dopiero po latach uszkodzenie nerek objawi się objawowo.
Na etapie białkomoczu amyloidoza nerek trwa 10 lat lub dłużej. W tym czasie amyloidoza stopniowo odkłada się w naczyniach, przestrzeni międzykomórkowej i kłębuszkach nerkowych. Z tego powodu nefrony odpowiedzialne za powstawanie moczu ulegają kompresji, zanikają i obumierają. Integralność filtra nerkowego, który normalnie nie przepuszcza dużych białek cząsteczkowych i krwinek, zostaje zerwana. Następnie białka są wydalane z moczem. Na tym etapie trudno podejrzewać amyloidozę nerek, ponieważ funkcja wydalnicza nie jest zaburzona. Problem można znaleźć w wynikach badań laboratoryjnych.
Amyloidoza nerek w stadium nerczycowym objawia się dalszym niszczeniem filtra nerkowego. Z tego powodu duża ilość białka jest tracona z moczem, jego stężenie we krwi spada. Białka są częścią procesu utrzymywania krwi w naczyniach. Wraz ze spadkiem stężenia białek płyn dostaje się do tkanek, obrzęk pojawia się o każdej porze dnia, niezależnie od pozycji ciała. Ponadto postępuje amyloidoza nerek, obrzęk jest silnie zaznaczony. Płyn gromadzi się w otrzewnej, jamie opłucnej, worku serca. Ten etap trwa 4-6 lat.
Na etapie azotemicznym funkcjonuje tylko 25% całej objętości tkanki nerkowej. To nie wystarczy, aby usunąć szkodliwe toksyny, mocznik, dlatego ich stężenie rośnie. Obraz kliniczny niewydolności nerek objawia się następująco:
- zaburzenia oddawania moczu. Zamiast przepisanych 800 ml dziennie pacjent wydala mniej niż 50 ml moczu;
- zdrowie się pogarsza, pojawiają się osłabienie, zmęczenie;
- trawienie jest zaburzone, apetyt zanika, pojawiają się nudności i wymioty, suchości w ustach towarzyszy nieprzyjemny zapach;
- skóra staje się blada, sucha, stale swędzi;
- cierpi układ sercowo-naczyniowy, co powoduje arytmię, wzrasta ciśnienie krwi, możliwy jest wzrost mięśnia sercowego;
- mózg pod wpływem wysokiego stężenia kwasu moczowego ulega uszkodzeniu, pojawiają się bezsenność i zaburzenia pamięci, drażliwość, spadek zdolności umysłowych;
- spadek hemoglobiny i czerwonych krwinek prowadzi do anemii.
Uszkodzenie wątroby
Amyloidoza układowa często objawia się uszkodzeniem wątroby. Złogi amyloidu wywierają nacisk na drogi żółciowe, naczynia krwionośne i komórki wątroby, powodując upośledzenie funkcji narządów. Podkreślając zespoły amyloidozy, wskazują na wzrost wątroby, wyczuwalny podczas badania palpacyjnego.
Powierzchnia wątroby pozostaje gładka, nie ma bólu. W przypadku długiego przebiegu choroby rzadko rozwija się niewydolność wątroby, co wiąże się ze zdolnościami regeneracyjnymi narządu.
Amyloidoza wątrobowa objawia się objawami:
- Powiększenie wątroby.
- nadciśnienie wrotne. Zwykle krew z narządów wewnętrznych dostaje się do wątroby, gdzie jest oczyszczana, a następnie wraca do krwioobiegu. Podczas ściskania naczyń wątroby amyloidem wzrasta ciśnienie w żyłach narządów wewnętrznych. W rezultacie pojawiają się obrzęki nóg, biegunka z krwią, krwawienie z przewodu pokarmowego.
- Żółtaczka występuje rzadko, jedynie w przypadku ucisku dróg żółciowych przez złogi amyloidowe. Jeśli to jest powód, swędzenie będzie towarzyszyć żółtaczce.
Niewydolność serca
Amyloidoza serca rozwija się w pierwotnych i innych postaciach dziedzicznych. W wyniku złogów amyloidu w mięśniu sercowym i błonach serca zaburzone zostaje krążenie krwi, komórki mięśniowe obumierają.
Objawy choroby:
- niemiarowość;
- restrykcyjna kardiomiopatia;
- niewydolność serca.
Arytmia występuje na tle złogów amyloidu w mięśniu sercowym, co zakłóca przewodzenie impulsu nerwowego. W rezultacie komory serca kurczą się nierównomiernie, pojawia się arytmia. Pacjent odczuwa zawroty głowy, obserwuje się omdlenie. Z powodu upośledzonego dopływu krwi do mózgu możliwy jest śmiertelny wynik.
Kardiomiopatia restrykcyjna występuje na tle złogów amyloidu w mięśniu sercowym. W efekcie mięsień sercowy pogrubia się, staje się mniej rozciągliwy, co prowadzi do złego funkcjonowania komór serca. Obraz kliniczny choroby to zmęczenie, duszność, gwałtowny spadek ciśnienia krwi przy zmianie pozycji z pozycji poziomej na pionową.
W przypadku niewydolności serca krążenie krwi w organizmie jest zaburzone. Przejawia się to obrzękiem, dusznością. Niewydolność serca w amyloidozie nie podlega standardowemu leczeniu chorób sercowo-naczyniowych. Choroba postępuje szybko, prowadząc do śmierci w ciągu kilku miesięcy.
Uszkodzenie nadnerczy i śledziony
 Nadnercza to gruczoły znajdujące się na każdej nerce i odpowiedzialne za wydzielanie hormonów. Amyloidoza zaburza funkcjonowanie narządów poprzez zatrzymanie syntezy hormonów. Jeśli amyloid odkłada się w śledzionie, narząd powiększa się, co jest zauważalne podczas badania palpacyjnego.
Nadnercza to gruczoły znajdujące się na każdej nerce i odpowiedzialne za wydzielanie hormonów. Amyloidoza zaburza funkcjonowanie narządów poprzez zatrzymanie syntezy hormonów. Jeśli amyloid odkłada się w śledzionie, narząd powiększa się, co jest zauważalne podczas badania palpacyjnego.
Normalnie śledziona usuwa z krwiobiegu zdeformowane komórki, które utknęły w jej strukturze. Złogi amyloidu w śledzionie powodują zablokowanie zdrowych czerwonych krwinek, płytek krwi i białych krwinek.
W efekcie rozwija się anemia (ogólne osłabienie, bladość skóry, duszności), małopłytkowość (krwawienia z nosa, krwotoki skórne), leukopenia (podatność na infekcje).
Uszkodzenie przewodu pokarmowego
Amyloidoza jelitowa może być uogólniona, gdy upośledzone jest wchłanianie składników odżywczych, i miejscowa, gdy nagromadzenia amyloidu imitują guz. W pierwszym przypadku pojawiają się objawy takie jak biegunka, utrata masy ciała, osłabienie, zaburzenia psychiczne, anemia. W drugim przypadku choroba charakteryzuje się zaparciami, bólami brzucha, wzdęciami.
Uszkodzenie stawów i mięśni
Amyloid najpierw atakuje małe stawy na stopach, dłoniach, w miarę postępu choroby osiada w łokciach i kolanach. Choroba charakteryzuje się bólem podczas ruchu, obrzękiem tkanki i zaczerwienieniem skóry, gorączką w dotkniętym obszarze, dysfunkcją stawu.
Amyloid odkłada się niepostrzeżenie przez długi czas w tkance łącznej, nie naruszając struktury mięśniowej i nie pojawiając się. Z biegiem czasu komórki tkanki mięśniowej są ściskane, dopływ krwi do nich jest zaburzony i obumierają. Choroba charakteryzuje się osłabieniem mięśni, bólem, stwardnieniem i przerostem mięśni.
Rozpoznanie amyloidozy
Rozpoznanie takie jak amyloidoza mogą podejrzewać lekarze różnych specjalności – reumatolodzy, kardiolodzy i urolodzy, neurolodzy, dermatolodzy itp. Dlatego rozpoznanie amyloidozy powinno opierać się na kompleksowej ocenie wywiadu, objawów klinicznych, badaniu laboratoryjnym i instrumentalnym . Aby zbadać stan narządów, zaleca się EKG, prześwietlenie przełyku, endoskopię, sigmoidoskopię. Jeśli podejrzewa się amyloidozę nerek, diagnoza koniecznie obejmuje USG jamy brzusznej.
Leczenie amyloidozy
Pomimo faktu, że istnieją różne poważne choroby, choroba amyloidozy niesie ze sobą złe rokowanie. Faktem jest, że we wczesnych stadiach nie można zidentyfikować choroby, a jej objawy kliniczne są zauważalne wiele lat po wystąpieniu choroby. Przy takiej diagnozie, jak amyloidoza nerek, leczenie jest jedynie wspomagające, ponieważ środki terapeutyczne nie są skuteczne.
Przy pierwszym podejrzeniu obecności choroby konieczna jest hospitalizacja w nefrologii w celu zbadania układu moczowo-płciowego, ponieważ uszkodzenie nerek jest uważane za najniebezpieczniejszy objaw. Inni specjaliści są również zaangażowani w badanie obecności uszkodzeń innych narządów.
Jeśli diagnoza nie wykazała poważnych naruszeń w funkcjonowaniu ważnych narządów, leczenie amyloidozy można przeprowadzić w domu, gdzie pacjent musi ściśle przestrzegać wszystkich zaleceń lekarskich. Leczenie może obejmować leki, dietę, dializy i przeszczepy narządów.
„Amyloidoza” to termin łączący grupę chorób wyróżniających się szeroką gamą objawów klinicznych i charakteryzujących się zewnątrzkomórkowym odkładaniem się nierozpuszczalnych patologicznych białek włóknistych w narządach i tkankach. Ta patologia została po raz pierwszy opisana w XVII wieku. Bonnet - sago śledziona u pacjenta z ropniem wątroby. W połowie XIX wieku. Virchow użył botanicznego terminu „amyloid” (z greckiego amylon, skrobia) do opisania materiału pozakomórkowego znalezionego w wątrobie podczas sekcji zwłok, ponieważ uważał, że ma on podobną strukturę do skrobi. Następnie ustalono białkowy charakter złogów, ale termin „amyloid” zachował się do dziś.
w latach 20. W XX wieku Benhold zaproponował barwienie amyloidu czerwienią Kongo, po czym odkryto efekt podwójnego załamania światła spolaryzowanego – zmianę ceglastej czerwieni na jabłkową. W 1959 roku Cohen i Calkins ustalili włóknistą strukturę amyloidu za pomocą mikroskopii elektronowej.
Kliniczne koncepcje amyloidozy również uległy ewolucji: Rokitansky w 1842 roku ustalił związek między „chorobą łojową” a gruźlicą, kiłą i riketsjozą; Wilks w 1856 roku opisał „narządy tłuszczowe” u pacjenta, który nie miał żadnych współistniejących chorób; Atkinson w 1937 roku odkrył amyloidozę u pacjentów ze szpiczakiem mnogim. Zidentyfikowano starcze (Soika, 1876) i dziedziczne (Andrade, 1952) formy choroby, amyloidozę podzielono na genetyczną, pierwotną i wtórną, a ostatecznie w 1993 roku przyjęto klasyfikację WHO, opartą na specyfice główne włókniste białko amyloidu.
W naszym kraju wielki wkład w rozwój idei dotyczących amyloidozy wnieśli E. M. Tareev, I. E. Tareeva, V. V. Serov. Ogromną rolę w badaniu pierwotnych i genetycznych wariantów amyloidozy i chorób okresowych odgrywa O. M. Vinogradova, której monografie opublikowane w 1973 i 1980 r. Nie straciły dziś na aktualności.
Obecnie amyloidozę dzieli się klinicznie na postać układową i miejscową. Wśród form systemowych, w zależności od składu złogów włóknistych, wyróżnia się cztery typy ( ).
Lokalne formy amyloidozy obejmują obecnie chorobę Alzheimera (A-beta, fibryle składają się z białka β odkładającego się w mózgu), amyloidozę wysp trzustkowych, prawdopodobnie mającą patogenetyczny związek z cukrzycą typu 2, amyloidozę występującą w guzach endokrynologicznych, guzy amyloidowe skóry, okolicy nosowo-gardłowej, pęcherza moczowego i innych rzadkich typów.
amyloidoza AL
Rozwój AL-amyloidozy jest możliwy w szpiczaku mnogim, chorobie Waldenströma, chłoniakach B-komórkowych i może być idiopatyczny w pierwotnej amyloidozie. Wszystkie te warianty łączy wspólna patogeneza, amyloidoza pierwotna jest najtrudniejsza do rozpoznania ze względu na brak wyraźnych objawów choroby hematologicznej, dlatego warto szczegółowo przyjrzeć się tej postaci.
W pierwotnej amyloidozie, łagodnej dyskrazji komórek plazmatycznych związanej ze szpiczakiem mnogim, nieprawidłowe klony komórek plazmatycznych szpiku kostnego wytwarzają amyloidogenne immunoglobuliny. Niektóre aminokwasy w regionach zmiennych łańcuchów lekkich tych immunoglobulin zajmują nietypową pozycję, co prowadzi do ich niestabilności i powoduje skłonność do fibrylogenezy. U pacjentów z pierwotną amyloidozą zawartość komórek plazmatycznych w szpiku kostnym wzrasta do 5-10% (normalnie mniej niż 4%, przy szpiczaku mnogim - ponad 12%) i wytwarzają one określony izotyp łańcuchów lekkich immunoglobulin, który dominuje w barwieniu immunohistochemicznym. Wolne monoklonalne łańcuchy lekkie dominującego izotypu lambda lub (rzadziej) kappa są wykrywane we krwi iw moczu, ale ich zawartość jest mniejsza niż w szpiczaku mnogim.
Obraz kliniczny amyloidozy pierwotnej jest różnorodny i uwarunkowany dominującym udziałem w procesie patologicznym określonych narządów – serca, nerek, układu nerwowego, przewodu pokarmowego, wątroby itp. Pierwszymi objawami są osłabienie i utrata masy ciała, ale na tym etapie etapie, przed pojawieniem się objawów narządowych, diagnoza jest niezwykle rzadka.
Najczęstszymi narządami docelowymi w amyloidozie AL są nerki i serce. Uszkodzenie nerek objawia się zespołem nerczycowym, uporczywym, a wraz z początkiem przewlekłej niewydolności nerek, krwiomoczem i nadciśnieniem tętniczym nie są typowe.
Wraz z odkładaniem się amyloidu w mięśniu sercowym rozwijają się różnorodne zaburzenia rytmu, postępująca niewydolność serca, którą mogą poprzedzać bezobjawowe zmiany w zapisie EKG w postaci spadku napięcia zębów. W badaniu echokardiograficznym stwierdza się koncentryczne pogrubienie ścian lewej i prawej komory, zmniejszenie objętości jam serca, umiarkowane zmniejszenie frakcji wyrzutowej oraz dysfunkcję rozkurczową mięśnia sercowego lewej komory.
Często występują objawy zajęcia układu nerwowego – autonomicznego, w postaci niedociśnienia ortostatycznego, oraz obwodowego – w postaci zaburzeń czucia. W ostatnich latach opisywano również zmiany w ośrodkowym układzie nerwowym, choć wcześniej uważano, że nie są one charakterystyczne dla pierwotnej amyloidozy.
Zjawiska dyspeptyczne (uczucie pełności, zaparcia, biegunki) i zespół złego wchłaniania mogą być spowodowane zarówno uszkodzeniem autonomicznego układu nerwowego, jak i amyloidozą przewodu pokarmowego. Bardzo charakterystyczna jest hepatomegalia, której charakter należy różnicować między przekrwieniem spowodowanym niewydolnością serca a amyloidowym uszkodzeniem wątroby. To ostatnie potwierdza wzrost poziomu fosfatazy alkalicznej w surowicy krwi. Śledziona jest często zajęta, ale splenomegalia nie zawsze jest wykrywana i nie ma wielkiego znaczenia klinicznego.
Makroglossia, klasyczny objaw pierwotnej amyloidozy, występuje u 20% pacjentów, naciek tkanek miękkich może prowadzić do zaniku mięśni i skóry, dystrofii paznokci, łysienia i pojawienia się guzopodobnych formacji – amyloidu.
Rzadziej występują uszkodzenia naczyniowe, których objawami są plamica okołooczodołowa – „oczy szopa pracza” oraz wybroczyny. Mogą wystąpić krwawienia, w tym krwawienia z pęcherza moczowego, spowodowane zarówno zmianami w ścianie naczyń, jak i naruszeniem układu krzepnięcia, przede wszystkim niedoborem czynnika X, który wiąże się z amyloidem. Charakterystyczną dla amyloidozy trombocytozę zwykle tłumaczy się niedoborem czynników krzepnięcia.
Amyloidozę płuc często stwierdza się tylko podczas sekcji zwłok. Jednak w niektórych przypadkach duszność, krwioplucie i wysięk opłucnowy mogą być spowodowane nie tylko zastoinową niewydolnością serca i zespołem nerczycowym, ale także amyloidową chorobą płuc. Możliwe jest odkładanie się amyloidu w pęcherzykach płucnych i rozwój amyloidoma płuc. Radiologicznie można wykryć zmiany siatkowe i guzkowe w tkance płucnej.
Zajęcie nadnerczy może prowadzić do niewydolności nadnerczy, która często pozostaje nierozpoznana, ponieważ niedociśnienie i hiponatremia są postrzegane jako objawy niewydolności serca i uszkodzenia autonomicznego układu nerwowego. U 10-20% pacjentów niedoczynność tarczycy może wystąpić jako przejaw uszkodzenia tarczycy, często dochodzi do powiększenia ślinianek podżuchwowych.
Rozpoznanie pierwotnej amyloidozy, oprócz wskazanych cech klinicznych, które mogą być podobne w amyloidozie wtórnej, opiera się na szeregu danych laboratoryjnych. U 85% pacjentów immunoelektroforeza białek surowicy krwi i moczu ujawnia obecność immunoglobulin monoklonalnych. W rutynowych badaniach te same monoklonalne immunoglobuliny znajdują się w moczu w postaci białka Bence-Jonesa. Biopsja szpiku kostnego umożliwia diagnostykę różnicową ze szpiczakiem mnogim, a także wykrywa umiarkowany wzrost liczby komórek plazmatycznych i ich monoklonalność za pomocą barwienia immunohistochemicznego.
Jednak nawet połączenie charakterystycznego obrazu klinicznego z obecnością monoklonalnych komórek plazmatycznych i białek jest wciąż niewystarczające do potwierdzenia rozpoznania pierwotnej amyloidozy. Dane z biopsji odgrywają tutaj decydującą rolę. Najmniej inwazyjna jest aspiracja podskórnej tkanki tłuszczowej przedniej ściany brzucha, która daje 80-90% pozytywnych wyników w AL-amyloidozie (metoda ta nie była dotychczas stosowana w naszym kraju). Biopsja dziąseł i błony śluzowej odbytnicy ma pewną wartość diagnostyczną, ale odsetek wyników dodatnich jest bardzo zróżnicowany w zależności od etapu procesu, dlatego wskazane jest wykonanie biopsji jednego z zajętych narządów – nerki, wątroby , serca, co daje prawie 100% dodatnie wyniki w amyloidozie typu AL.
Przede wszystkim materiał biopsyjny jest barwiony czerwienią Kongo. W przypadku wykrycia kongofilii badanego materiału konieczne jest zbadanie go w świetle spolaryzowanym, efekt dwójłomności jest charakterystyczny tylko dla amyloidu, inne substancje kongofilne nie uzyskują zielonego koloru jabłka. Następnie pożądane jest typowanie amyloidu. Najdokładniejsza jest metoda immunohistochemiczna wykorzystująca przeciwciała monoklonalne do białek prekursorowych amyloidu. Jednak obecnie w naszym kraju jest praktycznie niedostępny. Dlatego do diagnozy stosuje się barwienie roztworami zasadowej guanidyny lub nadmanganianu potasu, co pozwala, choć pośrednio, określić rodzaj złogów włóknistych.
Rokowanie w przypadku pierwotnej amyloidozy jest gorsze niż w przypadku innych postaci choroby, średnia długość życia nie przekracza dwóch lat, przy obecności choroby serca lub zmian wielonarządowych nieleczonych pacjenci umierają w ciągu kilku miesięcy. Najczęstszymi przyczynami zgonów są niewydolność serca i nerek, posocznica, powikłania naczyniowe i kacheksja. Podobieństwo patogenetyczne ze szpiczakiem mnogim pozwala liczyć na zahamowanie progresji choroby podczas chemioterapii, którą przeprowadza się w celu supresji monoklonalnych komórek plazmatycznych. Istnieje kilka schematów leczenia ().
Zastosowanie chemioterapii w przypadku skutecznego leczenia może wydłużyć życie chorych o okres od 10 do 18 miesięcy. Jednak skuteczność terapii jest niska, w szczególności ze względu na fakt, że w wielu przypadkach progresja choroby prowadzi do śmierci pacjentów przed zakończeniem leczenia, a także z powodu rozwoju cytopenii, zakaźnej powikłania, śmiertelne arytmie w leczeniu ultrawysokimi dawkami deksazonu. Zastosowanie dużych dawek melfolanu przy transplantacji autologicznych komórek macierzystych pozwala na uzyskanie remisji w ponad 50% przypadków, jednak stosowanie tej metody jest ograniczone ciężkością choroby, wiekiem pacjentów oraz zaburzeniami czynnościowymi narządu ruchu. serce i nerki. W wielu przypadkach możliwa jest jedynie objawowa terapia wspomagająca.
Amyloidoza AA
Do rozwoju amyloidozy AA dochodzi podczas przewlekłych procesów zapalnych, prekursorami amyloidu AA są białka ostrej fazy surowicy, α-globuliny wytwarzane przez komórki różnych typów, głównie neutrofile i fibroblasty. Wtórna amyloidoza rozwija się w reumatoidalnym zapaleniu stawów, chorobie Bechterewa, łuszczycowym zapaleniu stawów, różnych nowotworach, chorobie Hodgkina, wrzodziejącym zapaleniu jelita grubego i chorobie Leśniowskiego-Crohna, chorobach okresowych (rodzinna gorączka śródziemnomorska), a także gruźlicy, zapaleniu kości i szpiku, rozstrzeniach oskrzeli.
Charakterystycznymi cechami klinicznymi amyloidozy AA są u większości pacjentów uszkodzenia nerek, a także stosunkowo rzadko uszkodzenia wątroby i/lub śledziony (około 10%) oraz serca (wykrywane jedynie w badaniu echokardiograficznym). Makroglossia nie jest typowa dla wtórnej amyloidozy. Rozpoznanie stawia się na podstawie współistnienia amyloidozy nerkowej i przewlekłej choroby zapalnej, co potwierdza immunohistochemiczne barwienie materiału biopsyjnego, w naszym kraju stosuje się wspomniane już metody barwienia pośredniego.
Rokowanie w dużej mierze zależy od charakteru choroby podstawowej, w naturalnym przebiegu u jednej trzeciej pacjentów rozwija się niewydolność nerek po 5 latach od wykrycia białkomoczu. W przypadku choroby okresowej pięcioletnia przeżywalność wynosi 25%.
Leczenie polega na stłumieniu ogniska - źródła produkcji białek prekursorowych surowicy. Usunięcie guzów, sekwestrektomia, resekcja jelita, leczenie gruźlicy, zmniejszenie aktywności reumatoidalnego zapalenia stawów (z zastosowaniem cytostatyków) prowadzą do zahamowania progresji amyloidozy, a niekiedy do cofnięcia rozwoju objawów klinicznych, zwłaszcza nerczycowych zespół.
Stosowanie kolchicyny w chorobach okresowych jest metodą z wyboru, jej skuteczność została udowodniona, leczenie zapobiega rozwojowi amyloidozy i spowalnia jej postęp. W innych postaciach amyloidozy wtórnej skuteczność kolchicyny nie została potwierdzona.
Starcze i dziedziczne formy amyloidozy układowej, jak również postaci miejscowe, są rzadkie, amyloidoza dializacyjna jest dobrze znana specjalistom, w ogólnej praktyce prawie nigdy się z nią nie spotyka.
Leczenie objawowe nie zależy od rodzaju amyloidozy, ale od zajętego narządu docelowego ( ).
Amyloidoza, zwłaszcza pierwotna, jest uważana za rzadko występującą patologię, ale w rzeczywistości jest nie tyle rzadka, co trudna do zdiagnozowania. Właściwe rozpoznanie wymaga nie tylko znajomości kliniki i patogenezy tej choroby, ale także dostępności określonych możliwości diagnostycznych. Aby zilustrować ten punkt, przedstawiamy własne dane ( ). W Oddziale Nefrologii Miejskiego Szpitala Klinicznego w Moskwie im. S.P. Botkina w latach 1993-2003. Obserwowano 88 pacjentów, u których rozpoznano amyloidozę.
Rozpoznanie potwierdzono morfologicznie u wszystkich pacjentów z AL-amyloidozą, amyloidozą starczą i nieokreśloną oraz u 30 pacjentów z amyloidozą wtórną – łącznie 53 przypadki. Biopsję nerki wykonano u 12 chorych, biopsję wątroby u 2 chorych, jelit u 8 chorych, dziąseł u 12 chorych, aw 19 przypadkach rozpoznanie potwierdzono badaniem morfologicznym materiału skrawkowego.
W większości przypadków rozpoznanie amyloidozy ustalono po raz pierwszy w wyniku badania w oddziale nefrologii. Porównaliśmy diagnozy referencyjne i kliniczne wśród pacjentów z AL-amyloidozą ( ).
Tylko w dwóch przypadkach na 20 (10%) rozpoznanie skierowania brzmiało „skrobiawica pierwotna” i u jednego z tych pacjentów zostało postawione w Poradni Terapii i Chorób Zawodowych MMA, au drugiego w poradni zagranicznej.
Wszyscy chorzy, u których rozpoznano szpiczaka mnogiego z rozwojem AL-amyloidozy, zostali przeniesieni do oddziałów hematologii. Spośród 11 pacjentów z pierwotną amyloidozą, siedmiu pacjentów otrzymywało przerywaną chemioterapię za pomocą kombinacji melfolanu i doustnego prednizolonu, czterech z nich w połączeniu z leczeniem dializacyjnym, a inny pacjent otrzymał tylko dializę i leczenie objawowe. Spośród tych pacjentów pięciu zmarło w ciągu dwóch tygodni do dwóch lat od rozpoczęcia leczenia (wszyscy z powodu niewydolności nerek i uszkodzenia wielu narządów), jeden pacjent jest dializowany, jeden pacjent został skierowany na autologiczny przeszczep komórek macierzystych, a jeden pacjent jest leczony aż do czasu teraźniejszego. U jednego pacjenta chemioterapię opóźniono z powodu obecności długotrwałego, nie pozostawiającego blizny wrzodu żołądka, a dwóch kolejnych pacjentów odmówiło leczenia.
Wśród pacjentów z amyloidozą wtórną w naszym badaniu przeważali pacjenci z reumatoidalnym zapaleniem stawów, na drugim miejscu wśród przyczyn były przewlekłe zapalenie kości i szpiku oraz łuszczycowe zapalenie stawów, rzadziej występowały inne choroby ( ).
Leczenie reumatoidalnego zapalenia stawów i łuszczycowego zapalenia stawów prowadzono z zastosowaniem cytostatyków (metatreksat, azatiopryna), chociaż w wielu przypadkach możliwości terapii były ograniczone ze względu na obecność CRF i chorób współistniejących. Pacjenci z przewlekłym zapaleniem kości i szpiku kierowani byli na oddziały chirurgii ropnej. Pacjenci z chorobą Bechterewa i Leśniowskiego-Crohna otrzymywali specyficzne leczenie, chorzy na POChP i gruźlicę kierowani byli także do szpitali specjalistycznych. Jednego z pacjentów z guzem żołądka udało się skutecznie zoperować i w ciągu czterech lat obserwacji zespół nerczycowy stopniowo ustępował, w pozostałych przypadkach nowotworu rozpowszechnienie procesu pozwalało jedynie na leczenie objawowe, pacjent z limfogranulomatoza została przyjęta w stanie terminalnym. Śmiertelność wśród pacjentów z amyloidozą wtórną wynosiła 38% (z powodu pacjentów z zaawansowanymi zmianami w momencie rozpoznania). Wszyscy pacjenci z chorobą okresową otrzymywali terapię kolchicyną.
Cechy rozpoznania i zastosowania nowoczesnych metod leczenia amyloidozy pierwotnej można zilustrować następującym przykładem: pacjentka K., lat 46, została po raz pierwszy hospitalizowana pod koniec października 2002 roku z powodu obrzęków nóg, kołatania serca, braku miesiączki. Ma historię przeziębień, wycięcia wyrostka robaczkowego, dwóch porodów w trybie pilnym, oznak choroby nerek i żadnych chorób przewlekłych. W kwietniu 2002 roku zachorowała na ostre zapalenie górnego płata prawego płuca, była leczona ambulatoryjnie, otrzymywała iniekcje abaktalu i linkomycyny. Ze względu na lokalizację zapalenia płuc została przebadana w poradni gruźliczej, wykluczono rozpoznanie gruźlicy. Na początku czerwca po raz pierwszy pojawiły się obrzęki na nogach, pod kątem których nie była badana. Obrzęk po krótkim czasie samodzielnie wyeliminowany, a następnie wznowiony. Chora była hospitalizowana w szpitalu terapeutycznym, w badaniu stwierdzono białkomocz do 1,65%, hipoproteinemię (białko całkowite 52 g/l), ciśnienie krwi prawidłowe (120/80 mm Hg), osad moczu bez zmian, stężenie kreatyniny w osoczu również w normalnych granicach. Postawiono rozpoznanie „ostre kłębuszkowe zapalenie nerek”, wykonano leczenie ampicyliną, kurantami, heparyną, triampurem, wykonano wycięcie migdałków. Białkomocz utrzymywał się, obrzęk stopniowo narastał, dlatego w celu dalszego badania i leczenia pacjentkę z rozpoznaniem przewlekłego kłębuszkowego zapalenia nerek skierowano do szpitala. SP Botkin.
W badaniu skóra jest czysta, zabarwiona normalnie, anasarca, obrzęk masywny, gęsty, stwierdza się wodobrzusze, obwodowe węzły chłonne niepowiększone. BP 110/70 mm Hg. Art., tony serca są dźwięczne, wyraźne, rytmiczne, tętno 90 uderzeń/min, wątroba i śledziona niepowiększone, diureza do 1000 ml/dobę, stolec regularny, bez patologicznych zanieczyszczeń. W badaniu stwierdzono zespół nerczycowy - białkomocz 3 g/l, skąpy osad w moczu, hipodysproteinemia, hiperlipidemia (białko całkowite 39 g/l, albuminy 12 g/l, globuliny 7-30-15-19%, odpowiednio α 1 - α 2 -β-γ cholesterol 17,8 mmol/l, β-lipoproteiny 250 j.), przy analizie moczu na obecność białka Bence-Jonesa reakcja jest ujemna, dobowe wydalanie 17-KS nie jest zmniejszone. Kliniczne badanie krwi i inne parametry biochemiczne w normie, koagulogram - ciężka hiperfibrynogenemia, podwyższony poziom RKFM. Badanie immunoglobulin krwi: Ig-A - 0,35, Ig-M - 35,7 (dwie normy), Ig-G - 1,96 g / l. RTG klatki piersiowej, kości czaszki i miednicy, USG jamy brzusznej, nerek, tarczycy, ECHO-KG bez patologii, USG miednicy małej – cechy adenomiozy trzonu macicy, endoskopia – refluksowe zapalenie przełyku, Przewlekłe zapalenie żołądka. Podczas badania neuropatologa nie stwierdzono patologii, onkolog zdiagnozował mastopatię włóknisto-torbielowatą.
W celu wyjaśnienia genezy zespołu nerczycowego w znieczuleniu miejscowym pod kontrolą USG wykonano biopsję cienkoigłową nerki prawej, nie stwierdzono powikłań. W badaniu biopsji w mezangium kłębuszków nerkowych iw naczyniach pozakłębuszkowych odnotowuje się odkładanie się amyloidu. Ładunki amyloidu do 25% kłębuszkowych pętli naczyniowych. Badanie immunohistochemiczne nie wykazało specyficznej luminescencji. Gdy preparaty są traktowane alkalicznym roztworem guanidyny przez 2 godziny, kongofilia i ich właściwości w świetle spolaryzowanym są zachowane, co jest typowe dla AL-amyloidozy.
Aby wyjaśnić naturę amyloidozy AL, w laboratorium Immunotest przeprowadzono badanie immunochemiczne krwi i moczu. Ujawniono paraproteinemię M-lambda ze spadkiem poziomu immunoglobulin poliklonalnych i paraproteinurię typu lambda Bence-Jonesa na tle masywnego nieselektywnego białkomoczu. Chora była konsultowana przez hematologa, sugerowano obecność choroby Waldenströma i wykonano biopsję trepanem szpiku kostnego. Wniosek: w istniejących jamach szpiku kostnego widoczne są komórki wszystkich trzech pędów o prawidłowej hematopoezie oraz komórki limfoidalne nie tworzące skupisk. Rozpoznanie choroby Waldenströma odrzucono z powodu braku nacieku limfoidalnego w szpiku kostnym, powiększonych węzłów chłonnych i śledziony oraz braku podłoża guza.
Postawiono rozpoznanie amyloidozy pierwotnej z uszkodzeniem nerek, zespołem nerczycowym, zachowaną funkcją nerek, nie stwierdzono cech uszkodzenia innych narządów. Od stycznia 2003 r. rozpoczęto chemioterapię melfolanem 16 mg/dobę i prednizolonem 100 mg/dobę, cykle czterodniowe co sześć tygodni. Prowadzone jest również leczenie objawowe: furosemid, veroshpiron, preparaty potasu, famotydyna, transfuzje albumin. Do tej pory przeprowadzono pięć kursów chemioterapii z dobrą tolerancją, zmniejszyły się obrzęki, zmniejszył się białkomocz do 1,8 g/l, nieznacznie zmniejszyło się nasilenie hipodysproteinemii (białko całkowite 46 g/l, albuminy 18 g/l, α 2-globuliny 20%). Czynność nerek zachowana, stężenie kreatyniny w osoczu 1,3 mg/dl, w kontrolnych badaniach dynamicznych nie stwierdzono uszkodzeń innych narządów i układów.
Przypadek ten wyraźnie ilustruje fakt, że do rozpoznania amyloidozy niezbędne są badania morfologiczne, immunologiczne i immunochemiczne. Tak więc u naszego pacjenta najbardziej oczywistym rozpoznaniem klinicznym było „przewlekłe kłębuszkowe zapalenie nerek”, aw przypadku braku możliwości wykonania biopsji nerki takie rozpoznanie najprawdopodobniej zostałoby postawione. U pacjentki nie występowały kliniczne objawy ogólnoustrojowego charakteru choroby, przewlekłego procesu zapalnego, choroby układu krwionośnego, z wyjątkiem podwyższonego poziomu Ig-M. I dopiero dane uzyskane w badaniu biopsji nerki zaowocowały trepanobiopsją szpiku kostnego i badaniem immunochemicznym, które łącznie umożliwiły rozpoznanie pierwotnej amyloidozy przed pojawieniem się uszkodzeń ogólnoustrojowych. Rozpoczęto terapię patogenetyczną, co prawda na tle rozwiniętego już zespołu nerczycowego, ale przed wystąpieniem niewydolności nerek i jedynie 25% kłębuszków obciążonych amyloidem, co jest stosunkowo korzystne prognostycznie.
Podsumowując, zauważamy, że amyloidoza jest poważną chorobą o wysokiej śmiertelności, która jest niezwykle trudna do zdiagnozowania, jednak terminowe i wysokiej jakości badanie pacjentów umożliwia wcześniejsze zdiagnozowanie i terminowe zastosowanie odpowiedniej terapii, w z kolei pozwala na poprawę rokowania w tej grupie chorych.
Literatura
- Varshavsky V. A., Proskurneva E. P. Znaczenie i metody diagnozy morfologicznej amyloidozy we współczesnej medycynie // Praktyczna nefrologia. - 1998. - 2:16-23.
- Vinogradova OM Pierwotne i genetyczne warianty amyloidozy. — M.: Medycyna, 1980.
- Zakharova E. V., Khrykina A. V., Proskurneva E. P., Varshavsky V. A. Przypadek pierwotnej amyloidozy: trudności w diagnostyce i leczeniu // Nefrologia i dializa. - 2002. - 1:54-61.
- Rameev VV Osobliwości uszkodzenia nerek w AA i AL-amyloidozie: diss... cand. miód. Nauki. - M., 2003.
- Kozlovskaya L. V., Varshavsky V. A., Chegaeva T. V. i wsp. Amyloidoza: nowoczesne spojrzenie na problem // Nefrologia praktyczna. - 1998. - 2:24-26.
- Rodney H., Raymond L.C i Skinner M. // Układowe amyloidozy; New England Journal of Medicine, 1997. 337:898-909.
- Dhodapkar M.V., Jagannath S., Vesole d. i in. // Leczenie amyloidozy AL za pomocą deksametazonu i interferonu alfa / Leuc Lymphoma. 1997. - 27(3-4):351-365
- Gertz MA, Lacy MQ, Lust JA i wsp. // Badanie fazy II dużych dawek deksametazonu w leczeniu wcześniej leczonej amyloidozy łańcuchów lekkich immunoglobulin. Am J. Hematol, 1999, 61(2):115-119.
- Gertz MA, Lacy M.Q., Lust JA i wsp. // Badanie II fazy dużych dawek deksametazonu u nieleczonych pacjentów z pierwotną amyloidozą układową. Med Oncol 1999.-16(2):104-109
- Sezer O., Schmid P., Shweigert M. i wsp. // Szybkie odwrócenie zespołu nerczycowego z powodu pierwotnej ogólnoustrojowej amyloidozy AL po VAD i późniejszej chemioterapii wysokodawkowej z autologicznym wsparciem sprzedaży łodygi. Przeszczep szpiku kostnego. 1999. - 23(9): 967-969.
- Sezer O., Neimoller K., Jakob C. i in. // Nowe podejście do leczenia pierwotnej amyloidozy. Opinia eksperta Badacze Grugs. 2000. - 9(10):2343-2350
- Sezer O., Eucker J., Jakob C., Possinger K. // Diagnostyka i leczenie amyloidozy AL. Klinika Nefrol. 2000.-53(6):417-423.
- Skinner M. „Amyloidoza” Obecna terapia w alergii, immunologii i reumatologii. Książka Mosby-Year. 1996. - 235-240.
- Palladini G., Anesi E., Perfetti V. i in. Zmodyfikowany schemat podawania dużych dawek deksametazonu w przypadku pierwotnej amyloidozy układowej (AL). Brytyjski Dziennik Hematologii. 2001.-113:1044-1046.
E. V. Zacharowa
Moskiewski Miejski Szpital Kliniczny. S. P. Botkina
Tabela 2. Schematy leczenia pierwotnej amyloidozy
- Cykliczne podawanie doustne melfolanu (0,15-0,25 mg/kg mc./dobę) i prednizolonu (1,5-2,0 mg/kg mc./dobę) przez cztery do siedmiu dni co cztery do sześciu tygodni przez rok, aż do osiągnięcia dawki kursu 600 mg
- Doustne stosowanie melfolanu w dawce 4 mg/dobę przez trzy tygodnie, następnie po dwutygodniowej przerwie - 2-4 mg/dobę przez cztery dni w tygodniu nieprzerwanie do osiągnięcia dawki kursowej 600 mg, w połączeniu z prednizolon
- Dożylne podanie dużych dawek melfolanu (100-200 mg/m² powierzchni ciała przez 2 dni), a następnie przeszczep autologicznych komórek macierzystych
- IV deksametazon 40 mg przez cztery dni co trzy tygodnie przez osiem cykli
- Dożylne podanie deksametazonu w dawce 40 mg w 1-4, 9-12 i 17-20 dniu 35-dniowego cyklu, od 3 do 6 cykli, a następnie zastosowanie interferonu α w dawce 3- 6 milionów jednostek trzy razy dziennie w tygodniu
- Schemat winkrystyna-doksoribucyna-deksametazon (VAD).
