Oznaki leczenia heterotopii istoty szarej. Heterotopia podkorowa: lissencephaly
Słowa kluczowe: padaczka, ogniskowa dysplazja korowa, heterotopia istoty szarej, kortykografia
Cel: ocena wyników chirurgicznego leczenia padaczki u pacjentów z zaburzoną migracją neuronów w korze mózgowej.
Materiały i metody: operowany u 4 pacjentów w wieku od 20 do 37 lat (2 mężczyzn i 2 kobiety) z padaczką spowodowaną różnymi zaburzeniami rozwojowymi kory mózgowej.
wyniki: wszyscy pacjenci w obrazie klinicznym mieli napady częściowe z wtórnym uogólnieniem przez 6 do 22 lat przed hospitalizacją. MRI mózgu ujawniło ogniskową dysplazję korową u trzech pacjentów i rozlaną okołokomorową heterotopię istoty szarej mózgu u jednego pacjenta. Trzech pacjentów z FCD poddano implantacji elektrod kortykograficznych w celu określenia obszaru kory odpowiedzialnego za rozwój napadów padaczkowych. U chorych z FCD wykonano topektomię zmian za pomocą śródoperacyjnej kortykografii, chorego z heterotopią okołokomorową wykonano lobektomię prawego płata czołowego. Nie zaobserwowano powikłań infekcyjnych i neurologicznych po zabiegach. W badaniu morfologicznym preparatów stwierdzono u 2 pacjentów FCD typu Taylora, 1 pacjenta typu FCD nie-Taylora, au jednego rozlaną okołokomorową heterotopię istoty szarej. Po 12 miesiącach w okresie pooperacyjnym u trzech pacjentów z FCD wynik leczenia operacyjnego oceniono w klasie IA wg Engela (całkowite złagodzenie napadów), u pacjentki z heterotopią istoty szarej – II wg skali Engela (zmniejszenie częstości napadów o 50%)
wnioski. U pacjentów z padaczką lekooporną konieczne jest uwzględnienie możliwej etiopatogenetycznej roli zaburzeń migracji neuronów korowych. Leczenie chirurgiczne może być opcją uzyskania stabilnej remisji klinicznej i społecznej adaptacji pacjentów.
Schizencefalia to anomalia w strukturze kory. Występuje z powodu naruszenia rozwoju mózgu w 2-5 tygodniu ciąży. Choroba związana jest z upośledzoną migracją neuronów do kory mózgowej podczas tworzenia sieci neuronowych mózgu.
Zawartość:
Co to jest schizencefalia?
Z powodu niewystarczającego odżywiania naczyń lub jego braku część tkanki mózgowej nie powstaje. Schizencefalia nie jest procesem niszczenia tkanki, ale konsekwencją jej niedorozwoju (liniowy defekt w tkance mózgowej charakteryzujący się brakiem komórek istoty szarej).
Mediana wieku wystąpienia objawów wynosi 4 lata (zakres od 3 do 4 tygodni do 12 lat).
Schizencefalia jest dwojakiego rodzaju.
Szczelina zamknięta - 1 typ. Charakteryzuje się jednostronnym lub obustronnym liniowym przekrojem kory mózgowej o niedoskonałej budowie. Ściany szczelin zamykają się, komory komunikują się z przestrzenią podpajęczynówkową. Jama szczelinowa to mały rowek pokryty nabłonkiem wyściółkowym i oponami pajęczynówki. Nie jest wypełniony płynem mózgowo-rdzeniowym, dlatego niemożliwe jest zdiagnozowanie patologii w okresie prenatalnym rozwoju na neurosonografii.
Szczelina otwarta (otwarta) - typ 2. Można go zobaczyć z jednej lub obu stron. Ściany ubytku są oddzielone od siebie prześwitem wypełnionym płynem mózgowo-rdzeniowym. Jego długość: od ścian komór do przestrzeni podpajęczynówkowej. W badaniu ultrasonograficznym otwarta schizencefalia jest wykrywana przez wzrost komór.
Objawy
Zamknięta schizencefalia stanowi ponad 50% wszystkich zdiagnozowanych przypadków. W 30% przypadków choroba jest połączona z postępującym wodogłowiem, które jest eliminowane przez przetoki komorowe.Liczba i nasilenie objawów zależy od rodzaju schizencefalii: jednostronnej lub dwustronnej, od lokalizacji ubytku korowego.
Jednostronny rozszczepy powodują niedowład, częściowy lub całkowity paraliż po jednej stronie ciała. Większość dzieci w okresie dorastania ma przeciętne zdolności umysłowe, poziom sprawności fizycznej jest bliski normy.
Objawy jednostronnej schizencefalii zamkniętej u większości pacjentów ograniczają się do takich zaburzeń rozwojowych: brak inicjatywy, pozostawanie w tyle za dziećmi w równym wieku psychicznie i fizycznie (oczywiście podczas wspólnych zabaw), umiarkowane zaburzenia percepcji mowy. Zaburzenia w koordynacji ruchów obserwuje się po stronie ciała przeciwnej do dotkniętego obszaru.
dwustronny rozszczepy mają bardziej nasilone objawy: opóźnienia w rozwoju fizycznym i umysłowym, trudności w nauce języka i nauczaniu podstawowych przedmiotów w szkole. Ze względu na niedoskonałe połączenia między mózgiem a rdzeniem kręgowym możliwe jest ograniczenie funkcji motorycznych. Obustronna niekoordynacja jest możliwa w przypadku obustronnej (dwustronnej) schizencefalii, nawet przy niewielkich rozszczepach.
Inne oznaki schizenzfalii:
- niskie napięcie mięśniowe;
- wodogłowie (nagromadzenie płynu w komorach mózgu);
- małogłowie (głowa mniejsza niż normalnie), czasami wielkogłowie (z powodu wodogłowia);
- częste drgawki.
Obwód głowy noworodka w wieku poniżej jednego roku z wodogłowiem może wzrosnąć do 50-75 cm zamiast normalnych 40 cm po 3 miesiącach i 47 cm rocznie.
U wszystkich dzieci ze schizencefalią rozpoznaje się padaczkę ogniskową.(wyraźnie określona strefa epiaktywności).
Rodzaje napadów:
- Złożone napady ogniskowe - niewyraźna świadomość, odwrócenie głowy, nieruchoma fiksacja wzroku, mioklonie (drgawkowe drganie mięśni) kończyn dolnych. Zwykle widoczne tylko po jednej stronie ciała.
- Napady złożone z wtórnym uogólnieniem (poprzedzone aurą lub napadem ogniskowym).
- Proste napady.
Mniej powszechne są napady miokloniczne (rytmiczne drgania grup mięśni, które wywołują mimowolne ruchy) i toniczne (nieoczekiwane rozluźnienie mięśni). Mogą się powtarzać 4-8 razy w miesiącu lub rzadziej, czasami pojawiają się tylko kilka razy w życiu.
Częstotliwość i nasilenie napadów padaczkowych nie zależy od rodzaju schizencefalii, ale od obecności segmentów dysplazji korowej (nieprawidłowa struktura kory mózgowej).
W 100% przypadków schizencefalia charakteryzuje się naruszeniem wyższych funkcji korowych: wzrok, słuch, wrażliwość (zapach, dotyk, smak) o różnym nasileniu. Zaburzenia ruchowe są bardziej nasilone przy frontalnej lokalizacji rozszczepów.
Schizencefalia rzadko jest niezależną patologią. Zwykle wykrywany w połączeniu z grupą anomalii, powstały również w wyniku naruszeń procesów ontogenezy (rozwoju ciała) w czasie ciąży:
- dysgenezja(niedorozwój) lub brak ciała modzelowatego;
- komorowo-megalii(powiększenie komór z naruszeniem odpływu płynu mózgowo-rdzeniowego);
- hipoplazja móżdżku(odpowiedzialny za funkcje motoryczne i koordynację);
- polimikrogyria(dużo dodatkowych zwojów, nieprawidłowe ułożenie warstw kory mózgowej);
- heterotopia istoty szarej(nieprawidłowa akumulacja i nieprawidłowa lokalizacja);
- dylatacja(przemieszczenie) lub wady ścian, niedorozwój rogi komór mózgu.
Obraz kliniczny schizencefalii uzupełniają konsekwencje wady mózgu:
- wodogłowiem kształt głowy (nienormalnie wysokie czoło, powiększona górna część czaszki, prążki brwiowe silnie zarysowane i przesunięte do przodu, silnie zaznaczony wzór żylny na czole);
- naruszenia unerwienia mięśni, które zapewniają ruch gałek ocznych, wewnętrznych mięśni oka i powiek;
- nieprawidłowa mimika lub jej brak z powodu nieprawidłowego unerwienia mięśni twarzy;
- porażenie opuszkowe (zaburzenia mowy, połykania, niemożność kontrolowania (ruchu) mięśni twarzy);
- zwiększone napięcie mięśniowe;
- tetrapareza spastyczna (niedowład wszystkich kończyn, asymetria i upośledzenie napięcia mięśniowego);
- brak lub naruszenie odruchów bezwarunkowych;
Czasami neurologiczne objawy schizencefalii są mniej nasilone, niż lekarze początkowo podejrzewają na podstawie MRI.
Co powoduje schizencefalię?
Dokładna przyczyna schizencefalii jest nieokreślona. Większość badaczy przedstawia teorie związane z zaburzeniami genetycznymi i naczyniowymi.
Mutacje w genach homeobox odpowiedzialne za wzrost i migrację neuroblastów (protoplastów neuronów) są obserwowane u wielu dzieci ze schizencefalia, ale nie u wszystkich. Genetyczną teorię występowania potwierdzają przypadki schizencefalii u rodzeństwa.
Może to mieć wpływ na rozwój choroby infekcje (na przykład wirus cytomegalii) i leki .
Jakie procesy wywołują pojawienie się przestrzeni w istocie szarej?
Inni wyrażają odmienne zdanie: szczeliny w istocie szarej powstają w wyniku okluzja naczyń . Zablokowanie lub brak tętnic szyjnych wewnętrznych lub środkowych mózgu prowadzi do udaru niedokrwiennego, a następnie do martwicy mózgu.
Diagnostyka
Badanie i leczenie objawowe przeprowadzane są w oddziale psychoneurologicznym.Lekarze stosują następujące instrumentalne metody diagnostyczne:
- Rezonans magnetyczny.
- Rentgenowska tomografia komputerowa.
- Elektroencefalografię uzupełniają badania z otwieraniem i zamykaniem oczu, fotostymulacją i hiperwentylacją (dziecko proszone jest o szybki i głęboki wdech i wydech).
U wszystkich dzieci ze schizencefalią EEG wykazuje spowolnienie aktywności w tle, a także jedną z dwóch zmian:
- lokalna aktywność padaczkowa w rejonach czołowo-skroniowych;
- wszechobecna aktywność padaczkowa bez konkretnego skupienia.
Ze względu na obecność wodogłowia otwarta schizencefalia jest podobna do porencefalia jednak w drugim przypadku rozszczep nie jest pokryty tkanką nabłonkową, lecz tkanką łączną lub glejową (pomocniczą). Choroba może być mylona z holoprosencefalia (całkowity lub częściowy brak podziału przodomózgowia na półkule).
CT jest rzadko stosowany w diagnostyce schizencefalii, ponieważ MRI zapewnia pełniejszy obraz patologii.

Za pomocą rezonansu magnetycznego wykrywa się współistniejące zaburzenia rozwoju mózgu:
- heterotopia istoty szarej (guzki w istocie szarej pod wyściółką komór);
- hipoplazja nerwu wzrokowego (niedostateczna liczba aksonów, jednostek strukturalnych neuronów);
- agenezja przegrody przezroczystej w czołowej lokalizacji schizencefalii;
- dysplazja przegrodowo-oczna (zaburzenia rozwojowe przysadki mózgowej, przegrody przezroczystej, nerwu wzrokowego).
Leczenie
Zapewnia się leczenie objawowe schizencefalii.
Tetrapareza, niedowład połowiczy, drgawki, spastyczność mięśni, opóźnienie psychoruchowe, leczy się stymulacją elektryczną lub mikropolaryzacją mózgu, psychoterapię, leki przeciwpadaczkowe, terapię botulinową (blokowanie przekazywania niechcianych sygnałów z nerwów do mięśni), stosuje się leczenie ortopedyczne.
Pacjenci z łagodną schizencefalią nie doświadczają nawrotów po rozpoczęciu leczenia lekami przeciwpadaczkowymi.
Jacy lekarze poza neurologiem i neurochirurgiem pomogą dziecku?
Lekarze w co najmniej 3 specjalnościach mogą pomóc w poprawie jakości życia:
- Fizjoterapeuta zaleci terapię poprawiającą rokowanie rozwoju zdolności motorycznych, a mianowicie: umiejętność siedzenia i stania (w ciężkich przypadkach). Dzieci z łagodnymi objawami mogą odnieść korzyści z ćwiczeń wzmacniających mięśnie rąk i nóg.
- Usługi terapeuta zajęciowy będzie potrzebne, jeśli dziecko nie może wykonywać czynności wymagających dobrze rozwiniętych umiejętności motorycznych: jeść, ubierać się samodzielnie. Terapia zajęciowa umożliwi pełne życie i pełnienie funkcji w domu, przedszkolu, szkole.
- Logopeda poprawić umiejętności mówienia i połykania.
Jaka jest prognoza?
Schizencefalia ma przeważnie korzystne rokowanie na całe życie. W przypadku terminowego zapewnienia resuscytacji i / lub rehabilitacji oraz późniejszego leczenia następuje remisja. Problemy z aktywnością ruchową będą się utrzymywać przez całe życie, istnieje ryzyko upośledzenia umysłowego, ale większość pacjentów może w pełni żyć w społeczeństwie.Oprócz epilepsji głównym problemem u pacjentów ze schizencefalia jest wodogłowie. Przy stałym wzroście płynu z jednej strony dochodzi do przemieszczenia komór i ucisku otaczających tkanek, w tym rdzenia przedłużonego (reguluje czynność serca i czynność oddechową). Umiarkowane wodogłowie jest leczone farmakologicznie, ale nie zawsze lekarze mogą zaoferować inne opcje niż operacja pomostowania.
Historia małego pacjenta: chłopca, 2 lata.
Matka - 25 lat, ojciec - 29 lat, pierwsza ciąża, zadowalający stan zdrowia, brak szkodliwych czynników środowiskowych w miejscu zamieszkania i pracy.
Wodogłowie zostało po raz pierwszy zasugerowane przez USG w 34 tygodniu. Z poradni powiatowej pacjentka została skierowana do regionalnego ośrodka okołoporodowego.
Wielkość płodu w fetometrii odpowiadała wiekowi ciążowemu. Podczas badania mózgu w prawej półkuli stwierdzono ubytek z płynną zawartością. Zawarte w nim kłębuszki naczyniowe umożliwiły upewnienie się, że przyczyną jego powstania nie była torbiel. Poza otwartym kręgiem Willisa nie znaleziono żadnych innych zmian.
Postawiono diagnozę kliniczną: schizencefalia typu 2 (z otwartym rozszczepem). Po 5 tygodniach urodziło się dziecko płci męskiej. Waga: 3450 g, 7 punktów w skali Apgar. Bezpośrednio po urodzeniu wykonano NSG, diagnoza została potwierdzona. Matka i dziecko zostały wypisane ze szpitala czwartego dnia.
Minęły 2 lata. Dziecko pozostaje daleko w tyle za rówieśnikami w rozwoju psychomotorycznym (statyka, motoryka, reakcje sensoryczne, mowa, interakcje społeczne), zdolności motoryczne są ograniczone. Występuje zespół konwulsyjny i zmniejszenie odruchów kręgosłupa.
Obecność anomalii twarzoczaszki, które można zobaczyć wizualnie, ma negatywną wartość prognostyczną: małogłowie, wodogłowie kształt głowy. Podobne odchylenia mogą rozwinąć się u dziecka z otwartą schizencefalia.
Dziecko z zamkniętą schizencefalia otrzyma korzystną prognozę na całe życie. Otwarte szczeliny w istocie szarej, przeciwnie, prowadzą do opóźnienia rozwoju umysłowego lub psycho-mowy (ZPR lub ZPRR), zaburzeń ruchowych.
Historia dorosłego pacjenta: 20 lat.
Postępowanie w przypadku skarg na kręcz szyi (hałas i dzwonienie w uszach), napady padaczkowe z automatyzmami mowy (niekontrolowana wymowa słów), drgawki toniczno-kloniczne. Ataki padaczki prowadzą do utraty przytomności.
Od momentu urodzenia do przyjęcia do szpitala po ostatnim napadzie, który miał miejsce na zajęciach na uniwersytecie, nie zakładano rozpoznania schizencefalii.
Krótka anamneza. Przy urodzeniu nie zauważono żadnych nieprawidłowości, opóźnienie rozwojowe rozpoczęło się po 9 miesiącach, prawa strona nagle przestała być posłuszna. Po skontaktowaniu się z neurologiem dziecięcym zrobili rezonans magnetyczny i tomografię komputerową ze zdiagnozowanym mózgowym porażeniem dziecięcym (później okazało się, że diagnoza była błędna). Przepisano serię leków wazoaktywnych i neurometabolicznych, chociaż nie było odpowiednich wskazań.
Pierwszy napad padaczkowy wystąpił w wieku 8 lat. Następnie obserwowano drgawki z aurą słuchową i ciężkimi drgawkami, ale bez utraty przytomności. Przepisano wiele leków, w tym leki przeciwpadaczkowe, ale choroba postępowała.
Ostatnio ataki zaczęły się na kilka dni przed lub na początku miesiączki. W leczeniu padaczki przepisano kurs Depakine w połączeniu z Lamictalem. Liczba napadów spadła, ale jeśli się rozpoczęły, miało miejsce kilka napadów dziennie.
Wyniki diagnostyki podczas kontaktu z regionalnym szpitalem klinicznym. EEG wykazało umiarkowane zmiany aktywności bioelektrycznej, nieregularny rytm alfa, epiaktywność w okolicy skroniowej lewej półkuli. Obraz MRI jest charakterystyczny dla schizencefalii.
Wady wyglądu: zez rozbieżny, asymetria strefy nosowo-wargowej, podniebienie gotyckie (wysokie i wąskie, łukowate), złamany kształt łuków zębowych, rybia łuska (sucha, łuszcząca się skóra) w goleniach, skrócenie prawej ręki i nogi o 2 i 2,5 cm.
Problemy neurologiczne: astygmatyzm (częściowe rozmycie konturów obrazów, niewyraźne widzenie), po prawej stronie ciała wzrost odruchów ścięgnistych (skurcze mięśni podczas rozciągania), niedowład (spadek aktywności mięśni), zmniejszona wrażliwość. Niepewny w pozycji Romberga (stojąc prosto z wyciągniętymi ramionami). Polineuropatia (zmniejszona wrażliwość ramion poniżej łokcia, nadwrażliwość nóg poniżej kolan).
Pesymistycznie prognozuje się dzieci z padaczką lekooporną (to znaczy z napadami, których nie można kontrolować lekami). Obecność chorób współistniejących pogarsza jakość życia i zmniejsza dostępne możliwości.
Śmiertelny wynik jest możliwy w przypadku ostrych infekcji (w tym tych, które stały się przewlekłe), zaburzeń metabolicznych, ciężkiej zatrucia i niewydolności wielonarządowej.
Heterotopia subependymalna(heterotopia okołokomorowa) jest najczęstszą postacią heterotopii istoty szarej (SG), charakteryzującą się guzkami SG zlokalizowanymi bezpośrednio pod wyściółkami komór bocznych. Według morfologii można podzielić na:
- jednostronne ogniskowe
- dwustronna ogniskowa
- obustronnie rozproszony: falisty pasek SW otaczający komory.
Epidemiologia
Większość przypadków jest sporadyczna, niektóre są recesywne sprzężone z chromosomem X (Xq28). Kobiety mają stosunkowo łagodne upośledzenie funkcji poznawczych, a następnie rozwija się epilepsja. W przypadku chłopców dochodzi do poronienia samoistnego, zwykle z powodu wad rozwojowych układu sercowo-naczyniowego. Ocaleni są poważnie niepełnosprawni.
Obraz kliniczny
Najczęściej heterotopia subependymalna jest związana z padaczką i opóźnieniem rozwoju.
Patologia
Podobnie jak inne typy heterotopii, ten typ jest wynikiem naruszenia migracji neuronów. W niektórych przypadkach przyczyną rozwoju heterotopii podwyściółkowej jest naruszenie proliferacji komórek.
Guzki istoty szarej składają się ze skupisk neuronów i komórek glejowych. Warto zauważyć, że najczęściej znajdują się po prawej stronie, prawdopodobnie z powodu późniejszej migracji neuroblastów z prawej strony.
Przypadki sprzężone z chromosomem X wykazują mutacje w genie dla filaminy-1, białka, które sieciuje wewnątrzkomórkową aktynę. Ponadto, filamina-1 odgrywa również ważną rolę w rozwoju naczyń.
Diagnostyka
MRI jest metodą z wyboru, chociaż heterotopia okołokomorowa jest widoczna w TK i USG (jeśli rozmiar jest bardzo duży).
ultradźwięk
Guzki podwyściółkowe SW są zwykle hiperechogeniczne w porównaniu z prawidłową istotą białą i mogą również wystawać do światła komory (falowanie komór).
CT
W CT heterotopia podwyściółkowa pojawia się jako nieuwapniony obszar tkanki, który nie gromadzi środka kontrastowego, o gęstości podobnej do normalnej istoty szarej, wokół komór bocznych.
MRI
MRI przedporodowe
W późnej ciąży rozpoznanie podwyściółkowej heterotopii jest stosunkowo jasne. Przed 26 tygodniem ciąży obecność prawidłowej okołokomorowej macierzy zarodkowej kresomózgowia utrudnia wykrycie, podobnie jak ruchy płodu.
MRI poporodowe
W warstwie wyściółki obserwuje się drobne guzki istoty szarej, które zniekształcają kontur komór. Najczęściej lokalizacja znajduje się w okolicy trójkąta i rogów potylicznych. Inne obszary mózgu wydają się normalne.
Guzki istoty szarej są wizualizowane na wszystkich sekwencjach, także po kontrastach, gdzie, podobnie jak normalna istota szara, nie gromadzą środka kontrastowego.
Diagnoza różnicowa
- norma
- jądra ogoniaste
- wzgórze
- gwiaździak podwyściółkowy olbrzymiokomórkowy
- ma wyraźną akumulację kontrastu
- zlokalizowane w pobliżu otworu Monroe
- węzły podwyściółkowe w stwardnieniu guzowatym
- zwykle zwapniały (z wyjątkiem wczesnego dzieciństwa)
- wyższy sygnał T2 niż sygnał istoty szarej
- krwotok podwyściółkowy w badaniu ultrasonograficznym i przedporodowym MRI
- choć obraz może być podobny, badanie kontrolne w przypadku krwotoku determinuje ewolucję zmian
Jest to wynik zaburzeń w kształtowaniu się poszczególnych struktur mózgowych lub mózgu jako całości, które występują w okresie prenatalnym. Często mają niespecyficzne objawy kliniczne: głównie zespół padaczkowy, upośledzenie umysłowe i umysłowe. Nasilenie kliniki bezpośrednio koreluje ze stopniem uszkodzenia mózgu. Diagnozuje się je przedporodowo w USG położniczym, po urodzeniu – za pomocą EEG, neurosonografii i rezonansu magnetycznego mózgu. Leczenie objawowe: przeciwpadaczkowe, odwodnione, metaboliczne, psychokorektywne.
ICD-10
P00 P01 P02 P04

Informacje ogólne
Anomalie w rozwoju mózgu - wady, polegające na nieprawidłowych zmianach w budowie anatomicznej struktur mózgowych. Nasilenie objawów neurologicznych towarzyszących anomaliom mózgowym jest bardzo zróżnicowane. W ciężkich przypadkach wady rozwojowe są przyczyną przedporodowej śmierci płodu, stanowią do 75% zgonów wewnątrzmacicznych. Ponadto ciężkie anomalie mózgowe powodują około 40% zgonów noworodków. Czas wystąpienia objawów klinicznych może być inny. W większości przypadków anomalie mózgowe pojawiają się w pierwszych miesiącach po urodzeniu dziecka. Ponieważ jednak formowanie się mózgu trwa do 8 roku życia, wiele defektów pojawia się klinicznie już po 1. roku życia. W ponad połowie przypadków wady rozwojowe mózgu są połączone z wadami rozwojowymi narządów somatycznych. Prenatalne wykrywanie anomalii mózgu jest pilnym zadaniem praktycznej ginekologii i położnictwa, a ich diagnostyka i leczenie pourodzeniowe to priorytetowe zagadnienia współczesnej neurologii, neonatologii, pediatrii i neurochirurgii.
Powody
Najważniejszą przyczyną niepowodzeń rozwoju wewnątrzmacicznego jest wpływ na organizm kobiety w ciąży i na płód różnych szkodliwych czynników, które mają działanie teratogenne. Wystąpienie anomalii w wyniku dziedziczenia monogenowego występuje tylko w 1% przypadków. Za najbardziej wpływową przyczynę wad mózgu uważa się czynnik egzogenny. Wiele aktywnych związków chemicznych, skażenia radioaktywne i niektóre czynniki biologiczne mają działanie teratogenne. Nie bez znaczenia jest tu problem zanieczyszczenia środowiska ludzkiego, które powoduje wchłanianie toksycznych chemikaliów do organizmu kobiety w ciąży.
Różne efekty embriotoksyczne mogą być związane ze stylem życia samej kobiety w ciąży: na przykład palenie, alkoholizm, narkomania. Zaburzenia dysmetaboliczne u kobiety w ciąży, takie jak cukrzyca, nadczynność tarczycy itp., mogą również powodować anomalie mózgowe płodu. Wiele leków, które kobieta może przyjmować we wczesnym okresie ciąży, nieświadoma procesów zachodzących w jej ciele, ma również działanie teratogenne. Silne działanie teratogenne wywierają infekcje przenoszone przez kobietę w ciąży lub infekcje wewnątrzmaciczne płodu. Najbardziej niebezpieczne są cytomegalia, listerioza, różyczka, toksoplazmoza.
Patogeneza
Budowa układu nerwowego płodu zaczyna się dosłownie od pierwszego tygodnia ciąży. Już w 23. dniu ciąży tworzenie się końców cewy nerwowej, której niepełne zespolenie przedniego końca pociąga za sobą poważne anomalie mózgowe. Około 28 dnia ciąży powstaje przedni pęcherzyk mózgowy, który następnie dzieli się na 2 boczne, które stanowią podstawę półkul mózgowych. Ponadto powstaje kora mózgowa, jej zwoje, ciało modzelowate, struktury podstawowe itp.
Różnicowanie neuroblastów (zarodkowych komórek nerwowych) prowadzi do powstania neuronów tworzących istotę szarą i komórki glejowe, które tworzą istotę białą. Szara materia odpowiada za wyższe procesy aktywności nerwowej. W istocie białej istnieją różne ścieżki, które łączą struktury mózgowe w jeden funkcjonujący mechanizm. Noworodek urodzony o czasie ma taką samą liczbę neuronów jak osoba dorosła. Ale rozwój jego mózgu trwa, szczególnie intensywnie w pierwszych 3 miesiącach. życie. Następuje wzrost komórek glejowych, rozgałęzienia procesów neuronalnych i ich mielinizacja.
Awarie mogą wystąpić na różnych etapach powstawania mózgu. Jeśli wystąpią w ciągu pierwszych 6 miesięcy. ciąża mogą prowadzić do zmniejszenia liczby utworzonych neuronów, różnych zaburzeń w różnicowaniu i hipoplazji różnych części mózgu. W późniejszym czasie może nastąpić uszkodzenie i śmierć normalnie utworzonej substancji mózgowej.
Rodzaje anomalii mózgu
Bezmózgowie- Brak mózgu i akranii (brak kości czaszki). Miejsce w mózgu zajmują narośla tkanki łącznej i ubytki torbielowate. Mogą być pokryte skórą lub nagie. Patologia jest niezgodna z życiem.
przepuklina mózgowa- wypadanie tkanek i błon mózgowych przez ubytek kości czaszki z powodu jej niezamknięcia. Z reguły tworzy się wzdłuż linii środkowej, ale może być również asymetryczny. Mała przepuklina mózgowa może imitować krwiak głowonogowy. W takich przypadkach prześwietlenie czaszki pomaga ustalić diagnozę. Rokowanie zależy od wielkości i zawartości przepukliny mózgowej. Przy niewielkim występie i obecności ektopowej tkanki nerwowej w jej jamie skuteczne jest chirurgiczne usunięcie przepukliny mózgowej.
Małogłowie- zmniejszenie objętości i masy mózgu z powodu opóźnienia w jego rozwoju. Występuje z częstotliwością 1 przypadku na 5 tysięcy noworodków. Towarzyszy temu zmniejszony obwód głowy i nieproporcjonalny stosunek czaszki twarzy do mózgu z przewagą pierwszego. Małogłowie stanowi około 11% wszystkich przypadków upośledzenia umysłowego. W przypadku ciężkiej małogłowie jest możliwe idiotyzm. Często występuje nie tylko ZPR, ale także opóźnienie w rozwoju fizycznym.
Makrocefalia- wzrost objętości mózgu i jego masy. Znacznie rzadziej niż małogłowie. Makrocefalia zwykle łączy się z upośledzoną architekturą mózgu, ogniskową heterotopią istoty białej. Główną manifestacją kliniczną jest upośledzenie umysłowe. Może wystąpić zespół konwulsyjny. Występuje częściowa makrocefalia ze wzrostem tylko w jednej z półkul. Z reguły towarzyszy mu asymetria części mózgowej czaszki.
Torbielowata dysplazja mózgu- charakteryzuje się wieloma torbielowatymi jamami mózgu, zwykle połączonymi z układem komorowym. Torbiele mogą mieć różną wielkość. Czasami zlokalizowane tylko na jednej półkuli. Wiele torbieli mózgu występuje z padaczką, która jest oporna na leczenie przeciwdrgawkowe. Pojedyncze torbiele, w zależności od wielkości, mogą mieć przebieg subkliniczny lub towarzyszyć im nadciśnienie śródczaszkowe; często odnotowuje się ich stopniową resorpcję.
Holoprosencefalia- brak rozdzielenia półkul, w wyniku czego są one reprezentowane przez jedną półkulę. Komory boczne tworzą pojedynczą jamę. Towarzyszy gruba dysplazja czaszki twarzy i wady somatyczne. Martwe urodzenie lub śmierć odnotowuje się pierwszego dnia.
ogniskowa dysplazja korowa(FKD) - obecność w korze mózgowej obszarów patologicznych z gigantycznymi neuronami i nieprawidłowymi astrocytami. Ulubiona lokalizacja - skroniowe i czołowe obszary mózgu. Charakterystyczną cechą napadów padaczkowych w PKD jest obecność krótkotrwałych złożonych napadów z szybkim uogólnieniem, którym w początkowej fazie towarzyszą demonstracyjne zjawiska ruchowe w postaci gestów, deptania w jednym miejscu itp.
Heterotopia- nagromadzenia neuronów, na etapie migracji neuronów, opóźnione w drodze do kory. Heterotopy mogą być pojedyncze i wielokrotne, mieć kształt węzłowy i wstęgowy. Ich główną różnicą w stosunku do stwardnienia guzowatego jest brak zdolności do akumulacji kontrastu. Te anomalie w rozwoju mózgu objawiają się episyndromem i oligofrenią, których nasilenie bezpośrednio koreluje z liczbą i wielkością heterotopionów. W przypadku samotnej heterotopii napady padaczkowe zwykle pojawiają się po 10 roku życia.
Diagnostyka
Poważne anomalie mózgu można często zdiagnozować za pomocą oględzin. W pozostałych przypadkach ZPR, niedociśnienie mięśniowe w okresie noworodkowym oraz występowanie zespołu drgawkowego u dzieci w 1. roku życia pozwalają na podejrzenie anomalii mózgowej. Możliwe jest wykluczenie traumatycznego lub niedotlenionego charakteru uszkodzenia mózgu, jeśli nie ma danych dotyczących urazu porodowego noworodka, niedotlenienia płodu lub zamartwicy noworodka. Diagnostykę prenatalną wad rozwojowych płodu przeprowadza się za pomocą przesiewowego badania ultrasonograficznego w czasie ciąży. Ultradźwięki w pierwszym trymestrze ciąży mogą zapobiec narodzinom dziecka z poważną anomalią mózgu.
Jedną z metod wykrywania wad mózgu u niemowląt jest neurosonografia przez ciemiączko. Znacznie dokładniejsze dane u dzieci w każdym wieku i u dorosłych uzyskuje się za pomocą MRI mózgu. MRI umożliwia określenie charakteru i lokalizacji anomalii, wielkości torbieli, heterotopii i innych nieprawidłowych obszarów, w celu przeprowadzenia diagnostyki różnicowej z niedotlenieniem, urazami, guzami, zakaźnymi zmianami w mózgu. Rozpoznanie zespołu konwulsyjnego i wybór terapii przeciwdrgawkowej przeprowadza się za pomocą EEG, a także przedłużonego wideomonitoringu EEG. W przypadku rodzinnych przypadków anomalii mózgowych przydatna może być konsultacja z genetykiem z badaniami genealogicznymi i analizą DNA. W celu identyfikacji anomalii połączonych przeprowadza się badanie narządów somatycznych: USG serca, USG jamy brzusznej, radiografia narządów klatki piersiowej, USG nerek itp.
Leczenie anomalii mózgu
Terapia wad rozwojowych mózgu jest głównie objawowa, prowadzona przez neurologa dziecięcego, neonatologa, pediatrę, epileptologa. W przypadku zespołu konwulsyjnego wykonuje się terapię przeciwdrgawkową (karbamazepina, lewetyracetam, walproiniany, nitrazepam, lamotrygina itp.). Ponieważ padaczka u dzieci towarzysząca anomaliom rozwojowym mózgu jest zwykle oporna na monoterapię przeciwdrgawkową, przepisuje się kombinację 2 leków (na przykład lewetyracetam z lamotryginą). W przypadku wodogłowia przeprowadza się terapię odwodnienia, zgodnie ze wskazaniami, stosuje się operację pomostowania. W celu poprawy metabolizmu normalnie funkcjonujących tkanek mózgu, które w pewnym stopniu kompensują istniejącą wadę wrodzoną, możliwe jest przeprowadzenie kuracji neurometabolicznej z wyznaczeniem glicyny, witamin gr. B itp. Leki nootropowe stosuje się w leczeniu tylko w przypadku braku episyndromu.
Przy umiarkowanych i stosunkowo łagodnych anomaliach mózgowych zaleca się kompleksowe wsparcie psychologiczne dla dziecka, uczącego starsze dzieci w specjalistycznych szkołach. Techniki te pomagają zaszczepić umiejętności samoobsługi, zmniejszyć nasilenie upośledzenia umysłowego i, jeśli to możliwe, przystosować społecznie dzieci z wadami rozwojowymi mózgu.
Prognozowanie i zapobieganie
Rokowanie w dużej mierze zależy od ciężkości anomalii mózgowej. Niekorzystnym objawem jest wcześniejszy początek padaczki i jej odporność na trwającą terapię. Obecność współistniejącej wrodzonej patologii somatycznej komplikuje rokowanie. Skutecznym środkiem zapobiegawczym jest wykluczenie działania embriotoksycznego i teratogennego na kobietę w czasie ciąży. Planując ciążę, przyszli rodzice powinni pozbyć się złych nawyków, poddać się poradnictwu genetycznemu i przebadać pod kątem przewlekłych infekcji.

Główne morfologiczne części mózgu
- przodomózgowie (końcowy) mózg składa się z dwóch półkul mózgowych.
- Międzymózgowie składa się ze wzgórza, nabłonka, podwzgórza, przysadki mózgowej, która nie jest zawarta w międzymózgowiu, ale jest wyizolowana w osobny gruczoł.
- śródmózgowie składają się z nóg mózgu i podniebienia czworogłowego. Górne wzgórza dachu czworokąta są podkorowym ośrodkiem wzrokowym, a dolne wzgórza są podkorowym ośrodkiem słuchu.
- tyłomózgowie składa się z mostu i móżdżku.
- rdzeń. Połączenie rdzenia przedłużonego z rdzeniem kręgowym to otwór magnum.
Śródmózgowie, tyłomózgowie i rdzeń przedłużony są połączone w pień mózgu.

Wewnętrzna struktura półkul mózgowych.
- szare komórki
- Biała materia
Istota szara składa się z kory, która całkowicie pokrywa półkule mózgowe. Istota biała znajduje się pod szarą materią mózgu. Jednak obszary z szarą materią są również obecne w istocie białej - skupiskach komórek nerwowych. Nazywane są jądrami (jądrami). Zwykle istnieje wyraźna granica między istotą białą a szarą. Różnicowanie istoty białej i szarej jest możliwe w CT, ale lepiej różnicowane w MRI.

Dysplazja korowa
W dysplazji korowej granice między istotą białą a szarą są zatarte. W takim przypadku należy dodatkowo zastosować sekwencję inwersji odzyskiwania T1. Na tych obrazach widoczne będą granice, z wyjątkiem obszarów dysplazji korowej.

atak serca
W przypadku obrzęku cytotoksycznego, który rozwija się w pierwszych minutach zawału mózgu, zanika również różnicowanie istoty białej i szarej, co jest wczesnym objawem zawału mózgu w tomografii komputerowej.







Duże półkule mózgu
Półkule mózgu są oddzielone dużym procesem sierpowatym. Na każdej półkuli znajdują się 4 płaty:
- Płat czołowy.
- płat ciemieniowy
- płata potylicznego
Płat czołowy jest oddzielony od ciemieniowego centralnym lub ralandowym rowkiem, który jest doskonale widoczny zarówno na odcinkach osiowym, jak i strzałkowym.
Płat czołowy jest oddzielony od płata skroniowego bocznym rowkiem, który jest doskonale widoczny zarówno w przekroju strzałkowym, osiowym, jak i czołowym.
Płat ciemieniowy jest oddzielony od płata potylicznego bruzdą ciemieniowo-potyliczną o tej samej nazwie. Ta linia nadal oddziela baseny szyjne i podstawne.
Niektórzy autorzy przydzielają wyspę w osobnym rowku, który jest dużym obszarem kory pokrywającej wyspę z góry i z boku, tworzy wieczko (łac. pars opercularis) i jest uformowane z części sąsiednich płatów czołowych, skroniowych i ciemieniowych .
Podziel się granicami


Podziel się granicami
Granice płatów czołowych i ciemieniowych.
Omega -?
bruzda centralna
objaw wąsów- Zakręt postcentralny.
zakręt obręczy – zakręt postcentralny.
Aby poprawnie określić granicę płatów czołowych i ciemieniowych, najpierw znajdujemy bruzdę centralną. Symbol jest wpisany w ten rowek Omega -? na przekrojach osiowych.
Pomocny jest również objaw wąsa położonego prostopadle do linii pośrodkowej oraz obraz odpowiadający bruzdzie postcentralnej. Odpowiednio przed zakrętem postcentralnym znajduje się bruzda centralna.
Bruzda pasa.
Na odcinkach strzałkowych należy znaleźć ciało modzelowate nad nim znajduje się bruzda obręczy, która ciągnie się z tyłu i w górę do bruzdy zaśrodkowej, od której z przodu znajduje się bruzda centralna lub bruzda Rolanda.

Płat czołowy
Płat czołowy jest duży, a jednym z głównych zakrętów jest zakręt przedśrodkowy, który jest korowym centrum ruchu. W płacie czołowym odnotowuje się również zakręt wyższy, środkowy i dolny. Wymienione zwoje idą od góry do dołu i równolegle do siebie.
Na dolnej powierzchni płata czołowego znajdują się proste i oczodołowe zakręty, pomiędzy którymi znajdują się drogi węchowe i opuszki. Obszary te są uszkodzone przez uraz.

Urazowe uszkodzenie płata czołowego
U tego pacjenta odnotowujemy symetryczne uszkodzenia odcinków podstawnych obu płatów czołowych, co odpowiada zmianom pourazowym.

Obszar Broca
Ważnym obszarem jest również obszar Broca, który znajduje się w dystalnych częściach dolnego zakrętu czołowego. Jego lokalizacja jest ważna przy planowaniu interwencji neurochirurgicznych. Ta strefa jest łatwa do znalezienia, pamiętając ikonę McDonalda.

Zawał z zaangażowaniem w proces patologiczny obszar Broca
Ten pacjent ma ostry zawał z powodu niedrożności przedniej gałęzi M2 lewego MCA. Uszkodzenie płata czołowego z udziałem w procesie patologicznym okolicy Broki.

płat ciemieniowy
Za bruzdą centralną znajduje się zakręt postcentralny, który służy jako analizator korowy o wrażliwości ogólnej i proprioceptywnej.
Za nimi znajdują się górne i dolne płaciki ciemieniowe.
W płaciku ciemieniowym górnym znajduje się rdzeń analizatora skóry odpowiedzialnego za stereognozję - zdolność rozpoznawania obiektów za pomocą dotyku.
W dolnym płatku ciemieniowym znajduje się analizator motoryczny odpowiedzialny za apraksję - ruchy celowe i dobrowolne.
stereognozja- umiejętność rozpoznawania przedmiotów dotykiem.
Apraksja- naruszenie arbitralnych działań.

Zanik przedklinka
Zanik przedklinka jest wczesnym objawem choroby Alzheimera jeszcze przed zanikiem kory płatów skroniowych i hipokampa.
Przedklinek to odcinek płata ciemieniowego na wewnętrznej powierzchni obu półkul mózgowych, znajdujący się powyżej i przed ciałem modzelowatym.

płat skroniowy
W wydzielinie płata skroniowego
wyższy zakręt skroniowy
Środkowy zakręt skroniowy
Dolny zakręt skroniowy. Te trzy zwoje są do siebie równoległe i znajdują się w płaszczyźnie poziomej.
Zwoje Geschla znajdują się na powierzchni górnego zakrętu skroniowego. Są korowym ośrodkiem słuchu.
Zakręt przyhipokampowy znajduje się na dolnej powierzchni płatów skroniowych w rejonach przyśrodkowych. Haczyk wraz z hipokampem odpowiadają za zmysł węchu. Kiedy hipokamp jest uszkodzony, w pierwszej kolejności dochodzi do zaburzeń pamięci.
Obszar Wernickego. Obszar Wernickego znajduje się w dystalnych częściach górnego zakrętu skroniowego. Jest to strefa mowy sensorycznej.

Płata potylicznego
W płatach potylicznych określa się nieregularne bruzdy i zwoje, ale najbardziej stałą jest bruzda ostrogi zlokalizowana na przyśrodkowej powierzchni płata potylicznego. Wokół rowka ostrogi znajduje się 17, 18 i 19 pól Brodmanna, które stanowią korowy środek widzenia.

Okluzja PCA
Ten pacjent klinicznie zaobserwował zaburzenia widzenia spowodowane uszkodzeniem płata potylicznego, którego przyczyną był zawał serca (niedrożność PCA).
podkorowa istota szara







podkorowa istota szara
Podkorowa istota szara obejmuje:
- wzgórze
- jądra podstawowe
- jądro ogoniaste
- jądro soczewkowe, w którym izolowana jest skorupa i blada kulka.
- powłoka
Kapsuła wewnętrzna składa się z przedniej części uda, kolana i tylnej części uda.
Jak znaleźć tylne udo?
Pomiędzy wzgórzem a jądrem soczewkowym znajduje się ognisko hiperintensywne, które jest traktem piramidalnym. Z tego hiperintensywnego ogniska rysujemy linię do kolana, która będzie rzutem tylnej kości udowej torebki wewnętrznej.
NB - Nie myl tylnego kolana z bladą piłką.
Klasyfikując krwotoki śródmózgowe w podkorowej istocie szarej, w zależności od lokalizacji w stosunku do torebki wewnętrznej, krwotoki dzielą się na:
- boczny
- środkowy
- mieszany
BIAŁA MATERIA
Włókna spoidłowe łączące półkule.
Ciało modzelowate (największy komis)
Spoidło przednie
Spoidło tylne (spoidło sklepienia)

Spoidło przednie
Spoidło przednie znajduje się pod dziobem ciała modzelowatego za płytką końcową i łączy niektóre części mózgu węchowego: zakręt hipokampa, lewy i prawy haczyk płata skroniowego.
komisja tylna
Spoidło tylne należy do nabłonka, znajduje się u nasady nasady i łączy odpowiednie części śródmózgowia i międzymózgowia.
Wartość praktyczna:
Linia dwukomorowa w płaszczyźnie strzałkowej służy do oceny ciała modzelowatego. Linia dwukomorowa przebiega przez górną krawędź spoidła przedniego i dolną krawędź spoidła tylnego.

Ciało modzelowate
Ciało modzelowate składa się z:
Tułów lub ciało (przednie i tylne)
Każda sekcja łączy homolateralną sekcję mózgu.
Powstawanie ciała modzelowatego.
Ciało modzelowate rozwija się w specjalnej kolejności:
Od kolana, potem tułów, wałek i na końcu rozwija się dziób.
Mielinizacja ciała modzelowatego przebiega od tylnych do przednich regionów.
Ta wiedza pomaga zawęzić diagnostykę różnicową w patologiach ciała modzelowatego.

Dysgeneza i zanik ciała modzelowatego
W przypadku dysgenezy ciała modzelowatego kolano i przednie części ciała modzelowatego są dobrze uformowane, ale nie ma grzbietu i dzioba. Ta patologia jest wrodzona. Patologia jest pokazana po lewej stronie.
W przypadku atrofii ciała modzelowatego tylne części ciała modzelowatego (tylna część ciała i wałek) są dobrze uformowane, ale dziób, kolano i przednia część ciała są zmniejszone. Te zmiany są nabywane.
Wiele chorób dotyczy ciała modzelowatego, więc obecność zmian nie jest patognomoniczna dla konkretnej choroby.

Choroba Marchiafava-Bignami
Choroba Marchiafava-Bignami (centralne zwyrodnienie ciała modzelowatego, zespół Marchiafavy, mielinoliza pozamostowa).
Występuje u osób nadużywających alkoholu. U tych osób MRI ujawnia uszkodzenie grzbietu i tylnych części tułowia (ciała) ciała modzelowatego.
W przewlekłych stadiach choroby Marchiafava-Bignami ciało modzelowate jest wizualizowane w postaci kanapki, w której zachowane są górne i dolne warstwy ciała modzelowatego, ale z martwicą warstw środkowych.
Biała materia
Biała materia:
- okołokomorowy
- głębokie sekcje (ośrodki półkulowe)
- Włókna U
Istota biała okołokomorowa znajduje się w bliskiej odległości od komór bocznych mózgu.
Włókna U łączą korę pobliskich zakrętów lub podkorowej istoty białej.
Głębokie sekcje istoty białej zlokalizowane między okołokomorową i podkorową istotą białą.
Zmiany w istocie białej:
Zmiany istoty białej są klasyfikowane według lokalizacji:
- okołokomorowy
- przykorowy
- podkorowy
- zmiany w głębokiej istocie białej

Zmiany okołokomorowe
okołokomorowe (pojedyncze lub wielokrotne, małe lub duże, łączące się ze sobą)

Zmiany przykorowe
obok - ok. Ogniska te są zlokalizowane we włóknach U i przylegają bezpośrednio do istoty szarej, co oznacza, że między zmianą a istotą szarą nie ma warstwy istoty białej.
Kształtem te ogniska są różne, jak powtórzyć kształt włókien u, mogą być również zaokrąglone i nieregularne w kształcie. Ta lokalizacja jest patognomoniczna dla SM.

Zmiany podkorowe
Ogniska podkorowe to ogniska zlokalizowane w pobliżu kory mózgowej, ale jednocześnie między ogniskiem a korą znajduje się warstwa istoty białej.

Ogniska w głębokiej istocie białej.
Te ogniska występują w różnych chorobach mózgu.




KOMORY MÓZGU
Komory boczne składają się z:
- rogi przednie (przednie)
- rogi tylne (potyliczne)
- rogi dolne (czasowe)
Komory boczne są połączone z trzecią komorą przez sparowany otwór Monro.
Trzecia komora ma nieregularny kształt ze względu na obecność kieszonek. Otwarcie trzeciej komory odpowiada spoidłowi międzywzgórzowemu.
Trzecia komora jest połączona z czwartą komorą sylwiańskim akweduktem. Z czwartej komory płyn mózgowo-rdzeniowy wchodzi do cystern podstawnych przez sparowany otwór Luschki i niesparowany otwór Mogendi.
Przy ocenie komór warto zwrócić uwagę na rogi komorowe, gdyż w chorobach zwyrodnieniowych, takich jak choroba Alzheimera, zanikowi hipokampa towarzyszy poszerzenie rogów skroniowych. W trybie FLAIR sygnał z rogów tylnych (potylicznych) jest zwiększony, co jest normalne, podobnie jak asymetria rogów.
TRZECIA KOMORY.
Trzecia komora znajduje się w linii środkowej między guzkami wzrokowymi. Łączy się z komorami bocznymi przez otwory Monroe, a z czwartą komorą przez wodociąg mózgu.
Kieszenie trzeciej komory:
- nadskrzyżowaniowy
- lejkowaty
- Nadnerczy
- Szyszkowaty
Zwykle te kieszenie mają ostre rogi, ale wraz ze wzrostem nacisku kieszenie otwierają się.
Czwarta komora mózgu.
Czwarta komora jest jamą tyłomózgowia i za pomocą sparowanych otworów Luschki i niesparowanego otworu Magendie jest połączona z podstawowymi cysternami.

Sploty naczyniowe
Sploty naczyniówkowe wytwarzające płyn mózgowo-rdzeniowy zlokalizowane są we wszystkich komorach mózgu, tak więc zwapnienie splotu naczyniówkowego, które częściej uwidacznia się w rogach tylnych komór bocznych, jest widoczne zarówno w trzeciej, jak i czwartej komorze.

stwardnienie guzowate.
Zwapnienie splotów naczyniówkowych, które jest normą, nie powinno być mylone ze stanami patologicznymi. Na przykład ze zwapnieniami komór bocznych - bulw okołokomorowych w stwardnieniu guzowatym.

Heterotopowa istota szara
Należy pamiętać, że jedyną szarą materią graniczącą z komorami bocznymi są jądra ogoniaste, które mają wyraźne, równe kontury. Dodatkowe struktury istoty szarej, które deformują kontur komór bocznych, są zmianami patologicznymi charakterystycznymi dla heterotopii istoty szarej.

Warianty budowy komór
- wnęka przezroczystej przegrody, którą obserwuje się u większości noworodków (z czasem zamyka się) i wygląda jak trójkątny kształt między korpusami przedniej komory bocznej. Ta jama nigdy nie przecina otworu Monroe.
- wnęka żagla pośredniego. Jedna ze ścian wnęki, która tworzy dach trzeciej komory.
- Jama Verge'a to rozciągnięta jama pomiędzy korpusami komór bocznych.

torbiel koloidalna
Od torbieli koloidalnej należy odróżnić warianty strukturalne, które będą różnić się intensywnością sygnału z płynu mózgowo-rdzeniowego w prawie wszystkich sekwencjach impulsów. Po wprowadzeniu środka kontrastowego torbiele koloidalne nie gromadzą kontrastu, co odpowiada łagodnemu procesowi.

Norma MRI - mediana przekroju strzałkowego. CSF - zbiorniki.
A - ZBIORNIK Z PŁYTĄ KOŃCOWĄ
B - KAWAŁKA CHIASMA
C - spłuczka międzynasadowa
D - Zbiornik obejściowy
E - Czworokątna cysterna
F - Cysternomóżdżkowa cysterna
G - Cysternocerebellar cysterna Prepontine pontocerebellaris
H - BOCZNE CASTERNA MÓZGODŹDZKA
I - ZBIORNIK MAGNA
Zdjęcie dzięki uprzejmości dr. Coenraad J. Hattingh
PUSZKI MÓZGU
Z czwartej komory mózgu płyn mózgowo-rdzeniowy dostaje się do podstawowych cystern za pomocą sparowanych otworów Luschki i niesparowanego otworu Magendie.
Nazwa zbiorników na podstawie lokalizacji:
W płaszczyźnie strzałkowej:
- Cysterna nadsiodłowa
- Cysterna mostowa, przez którą przechodzi główna arteria.
- Cysterna z czterema wzgórzami
- Duża lub podstawna cysterna mózgu
W płaszczyźnie osiowej:
- Spłuczka międzynasadowa
- Spłuczka obejściowa łączy spłuczkę międzynasadową i czworokątną. Od zbiornika obejściowego odróżnia się również skrzydła: prawe i lewe.


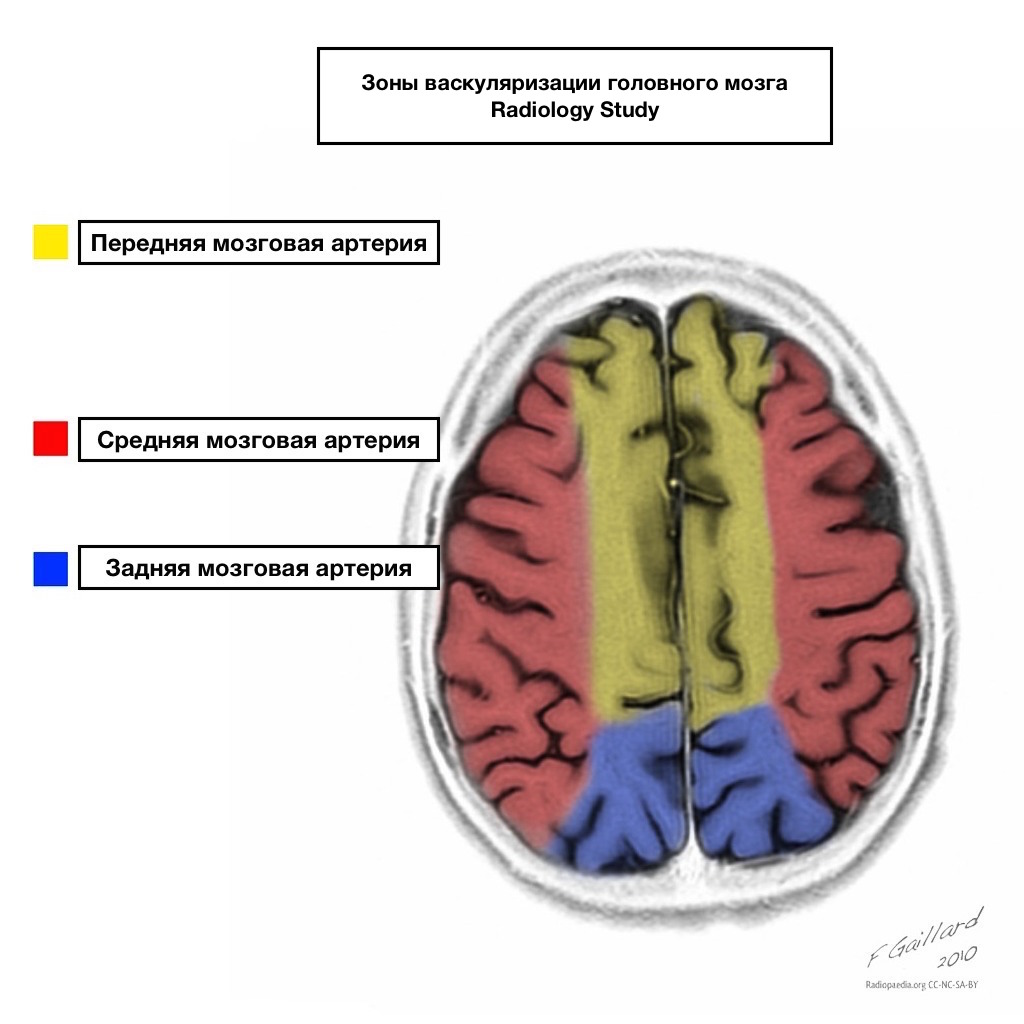







Pule dopływu krwi mają wyraźne granice.

Obszary sąsiedniego dopływu krwi
Strefy sąsiedniego dopływu krwi na przecięciu stref ukrwienia:
tętnica przednia mózgu
tętnica środkowa mózgu
Tętnica tylna mózgu.
Najczęściej zawały w tych obszarach mają charakter hemodynamiczny, to znaczy występują, gdy spada ciśnienie krwi.

Muszle mózgu
Mózg pokryty jest trzema błonami.
- Miękka skorupa jest ściśle przymocowana do mózgu, wchodzi we wszystkie pęknięcia i bruzdy, a w niej znajdują się naczynia krwionośne. W niektórych miejscach przenika do komór mózgu i tworzy splot naczyniówkowy.
- Błona pajęczynówki lub pajęczynówki leży nad bruzdami i rozprzestrzenia się od jednego zakrętu do drugiego.
- Twarda skorupa od wewnątrz wyściela wnęki czaszki, ściśle do nich przylega i tworzy zatoki żylne oraz wyrostki oddzielające od siebie poszczególne struktury mózgu.
Zwykle błony mózgu nie są wizualizowane na MRI, ale po wprowadzeniu kontrastu opona twarda jest kontrastowana.

Zmiany w miękkich oponach.
W rakowiaku opon miękkich na obrazach T1 i T2 bez kontrastu następuje wzrost sygnału z opon mózgowo-rdzeniowych, a po wprowadzeniu kontrastu poprawia się wizualizacja.

Zapalenie opon mózgowych
Zmiany w oponach często występują również w zmianach zapalnych, na przykład w gruźliczym zapaleniu opon mózgowo-rdzeniowych.

Zmiana Dura
Zmiana opony twardej występuje wraz z niedociśnieniem śródczaszkowym. Dzięki tej patologii uwidacznia się pogrubiona opona twarda, intensywnie gromadząca kontrast. Dodatkowymi kryteriami rozpoznania jest powiększenie przysadki mózgowej, wypadanie migdałków móżdżku do otworu wielkiego.

Zmiany w oponie twardej występują również w rakowiaku oponowo-pęcherzowym, które objawiają się pogrubieniem opony twardej z intensywnym nagromadzeniem środka kontrastowego i obrzękiem naczyniopochodnym sąsiednich części płata czołowego.
![]()
Przestrzenie powłoki.
Przestrzenie muszli to przestrzenie między muszlami mózgu.
- Przestrzeń podpajęczynówkowa to przestrzeń między pia mater a pajęczynówką. Normalnie powinien mieć intensywność płynu mózgowo-rdzeniowego.
- Przestrzeń podtwardówkowa to przestrzeń między pajęczynówką a oponą twardą.
- Przestrzeń nadtwardówkowa to przestrzeń między oponą a kośćmi czaszki, której normalnie nie widać, ponieważ opona jest zrośnięta z kośćmi czaszki.

Zmiana w przestrzeni podpajęczynówkowej
Zmiana w przestrzeni podpajęczynówkowej
Zwężenie. Zmiany te występują podczas ekspozycji wolumetrycznej (guz, zawał).
Rozbudowa. Zmiany te występują w okresie pourazowym, po zawale serca lub podczas atrofii.



Krwotoki muszlowe
W przypadku krwotoków muszlowych możemy doskonale zidentyfikować muszle.
Rodzaje krwotoków muszlowych:
krwotok nadtwardówkowy. Zwykle widziany jako soczewka i nie wystaje poza szwy, ale może przecinać zatoki mózgu, co różni się od krwotoków podtwardówkowych, które nigdy nie przechodzą przez zatoki mózgu.
Krwotok podtwardówkowy. Najczęstszą przyczyną jest pęknięcie żył powierzchownych w wyniku przemieszczenia mózgu podczas urazu. Jeśli w tym przypadku pęknie również błona podpajęczynówkowa, to w tym przypadku płyn mózgowo-rdzeniowy przedostanie się do przestrzeni podtwardówkowej.
Krwotok podpajęczynówkowy. Wykryto wzrost sygnału z płynu mózgowo-rdzeniowego w trybie FLAIR. Najczęstszą przyczyną krwotoku podpajęczynówkowego jest pęknięcie tętniaka, ponieważ tętnice zaopatrujące mózg są zlokalizowane w przestrzeni podpajęczynówkowej.

W procesach patologicznych w muszlach nie używa się terminu płaty, ale zamiast tego używa się terminu region. Na przykład ten pacjent ma oponiaka czołowego.
