Segni di trattamento dell'eterotopia della materia grigia. Eterotopia sottocorticale: lissencefalia
Parole chiave: epilessia, displasia corticale focale, eterotopia della sostanza grigia, corticografia
Bersaglio: valutazione dei risultati del trattamento chirurgico dell'epilessia in pazienti con disturbi della migrazione dei neuroni nella corteccia cerebrale.
Materiali e metodi: sono stati operati 4 pazienti di età compresa tra 20 e 37 anni (2 uomini e 2 donne) affetti da epilessia causata da vari disturbi dello sviluppo della corteccia cerebrale.
risultati: tutti i pazienti presentavano crisi parziali con generalizzazione secondaria nel quadro clinico da 6 a 22 anni prima del ricovero. La risonanza magnetica del cervello ha rivelato una displasia corticale focale in tre pazienti e un'eterotopia periventricolare diffusa della materia grigia del cervello in un paziente. Tre pazienti con FCD sono stati sottoposti a impianto di elettrodi corticografici per determinare l'area corticale responsabile dello sviluppo delle convulsioni. I pazienti con FCD sono stati sottoposti a topectomia delle lesioni con corticografia intraoperatoria, mentre i pazienti con eterotopia periventricolare sono stati sottoposti a lobectomia del lobo frontale destro. Dopo gli interventi non sono state notate complicazioni infettive o neurologiche. Uno studio morfologico dei campioni ha rivelato la FCD di tipo Taylor in 2 pazienti, la FCD di tipo non-Taylor in un paziente e l'eterotopia periventricolare diffusa della materia grigia in un paziente. Dopo 12 mesi nel periodo postoperatorio, in tre pazienti affetti da FCD, il risultato del trattamento chirurgico è stato valutato come classe IA sulla scala Engel (completa libertà da convulsioni), in un paziente con eterotopia della sostanza grigia - II sulla scala Engel (riduzione nella frequenza degli attacchi del 50%)
conclusioni. Nei pazienti con epilessia farmacoresistente è necessario tenere conto del possibile ruolo eziopatogenetico dei disturbi nella migrazione dei neuroni corticali. Il trattamento chirurgico può essere un’opzione per ottenere una remissione clinica stabile e l’adattamento sociale dei pazienti.
La schizencefalia è un'anomalia nella struttura della corteccia. Si verifica a causa di uno sviluppo cerebrale compromesso a 2-5 settimane di gravidanza. La malattia è associata a un'interruzione della migrazione dei neuroni verso la corteccia cerebrale durante la formazione delle reti neurali nel cervello.
Contenuto:
Cos'è la schizencefalia?
A causa dell'insufficiente nutrizione vascolare o della sua assenza, parte del tessuto cerebrale non si forma. La schizencefalia non è un processo di distruzione dei tessuti, ma una conseguenza del suo sottosviluppo (un difetto lineare nel tessuto cerebrale, caratterizzato dall'assenza di cellule della sostanza grigia).
L'età media di insorgenza dei sintomi della malattia è di 4 anni (da 3-4 settimane a 12 anni).
Esistono due tipi di schizencefalia.
Fessura chiusa - tipo 1.È caratterizzata da un'area lineare unilaterale o bilaterale della corteccia cerebrale con una struttura imperfetta. Le pareti delle fessure si chiudono, i ventricoli comunicano con lo spazio subaracnoideo. La cavità della fessura è un piccolo solco ricoperto da epitelio ependimale e midollo aracnoideo. Non è pieno di liquido cerebrospinale, quindi è impossibile diagnosticare la patologia nel periodo prenatale utilizzando la neurosonografia.
Fessura aperta (aperta) - tipo 2. Può essere osservato su uno o entrambi i lati. Le pareti del difetto sono separate l'una dall'altra da un lume pieno di liquido cerebrospinale. La sua lunghezza: dalle pareti dei ventricoli allo spazio subaracnoideo. All'ecografia, la schizencefalia aperta viene rilevata dall'allargamento dei ventricoli.
Sintomi
La schizencefalia chiusa rappresenta oltre il 50% di tutti i casi diagnosticati. Nel 30% dei casi, la malattia è associata all'idrocefalo progressivo, per eliminare il quale viene eseguito lo shunt ventricolare.Il numero e la gravità dei sintomi dipendono dal tipo di schizencefalia: unilaterale o bilaterale e dalla localizzazione del difetto corticale.
Unilaterale le fessure causano paresi, paralisi parziale o completa su un lato del corpo. La maggior parte dei bambini, quando cresce, ha capacità mentali nella media e il livello delle capacità fisiche è vicino alla norma.
I segni di schizencefalia chiusa unilaterale nella maggior parte dei pazienti sono limitati ai seguenti disturbi dello sviluppo: mancanza di iniziativa, ritardo mentale e fisico rispetto ai bambini della stessa età (evidente durante i giochi congiunti), disturbi moderati nella percezione del linguaggio. Si osserva una compromissione della coordinazione dei movimenti sul lato del corpo opposto all'area interessata.
Doppia faccia le schisi presentano sintomi più gravi: ritardi nello sviluppo fisico e mentale, difficoltà nell'apprendimento della lingua e nell'apprendimento delle materie di base a scuola. A causa delle connessioni imperfette tra cervello e midollo spinale, le funzioni motorie possono essere limitate. L'incoordinazione bilaterale è possibile nella schizencefalia bilaterale (bilaterale), anche con piccole fessure.
Altri segni di schizenfalia:
- tono muscolare basso;
- idrocefalo (accumulo di liquido nei ventricoli del cervello);
- microcefalia (testa più piccola del normale), talvolta macrocefalia (dovuta all'idrocefalo);
- convulsioni frequenti.
La circonferenza della testa di un neonato di età inferiore a un anno con idrocefalo può aumentare fino a 50-75 cm invece dei normali 40 cm a 3 mesi e 47 cm all'anno.
A tutti i bambini affetti da schizencefalia viene diagnosticata l’epilessia focale(zona di epiattività chiaramente limitata).
Tipi di attacchi:
- Crisi focali complesse: coscienza nebbiosa, rotazione della testa, sguardo fisso, mioclono (spasmi muscolari convulsivi) degli arti inferiori. Di solito si vede solo su un lato del corpo.
- Attacchi complessi con generalizzazione secondaria (preceduti da un'aura o attacco focale).
- Convulsioni semplici.
Meno comuni sono gli attacchi mioclonici (contrazioni ritmiche di gruppi muscolari, che provocano movimenti involontari) e tonici (rilassamento improvviso dei muscoli). Possono ripetersi 4-8 volte al mese o meno, a volte si verificano solo poche volte nella vita.
Frequenza e gravità delle crisi epilettiche dipende non dal tipo di schizencefalia, ma dalla presenza di segmenti di displasia corticale (struttura anormale della corteccia cerebrale).
Nel 100% dei casi, la schizencefalia è caratterizzata da una violazione delle funzioni corticali superiori: vista, udito, sensibilità (olfatto, tatto, gusto) di varia gravità. I disturbi motori sono più pronunciati nella localizzazione frontale delle schisi.
La schizencefalia è raramente una patologia indipendente. Di solito rilevato in combinazione con un gruppo di anomalie, formato anche a seguito di disturbi nei processi di ontogenesi (sviluppo del corpo) durante la gravidanza:
- disgenesia(sottosviluppo) o assenza del corpo calloso;
- ventricolomegalia(ventricoli dilatati con deflusso alterato del liquido cerebrospinale);
- ipoplasia cerebellare(responsabile delle funzioni motorie e della coordinazione);
- polimicrogiria(molte circonvoluzioni extra, disposizione errata degli strati della corteccia cerebrale);
- eterotopia della materia grigia(accumulo anomalo e localizzazione errata);
- dilatazione(spostamento) o difetti del muro, sottosviluppo corni dei ventricoli del cervello.
Il quadro clinico della schizencefalia è completato dalle conseguenze di un difetto cerebrale:
- forma della testa idrocefalica (fronte anormalmente alta, parte superiore del cranio allargata, arcate sopracciliari fortemente definite e spostate in avanti, disegno venoso fortemente pronunciato sulla fronte);
- disturbi nell'innervazione dei muscoli che forniscono il movimento dei bulbi oculari, dei muscoli interni dell'occhio e delle palpebre;
- espressioni facciali anormali o mancanza di esse dovute a innervazione impropria dei muscoli facciali;
- paralisi bulbare (difficoltà nel linguaggio, nella deglutizione, incapacità di controllare (muovere) i muscoli facciali);
- aumento del tono muscolare;
- tetraparesi spastica (paresi di tutti gli arti, asimmetria e disturbi del tono muscolare);
- assenza o compromissione dei riflessi incondizionati;
A volte i segni neurologici della schizencefalia sono meno gravi di quanto inizialmente ipotizzato dai medici sulla base dei risultati della risonanza magnetica.
Cosa causa la schizencefalia?
La causa esatta della schizencefalia non è chiara. La maggior parte dei ricercatori avanza teorie relative a disturbi genetici e vascolari.
Mutazioni nei geni homeobox , responsabili della crescita e della migrazione dei neuroblasti (precursori dei neuroni), sono osservati in molti, ma non in tutti i bambini affetti da schizencefalia. La teoria genetica dell'insorgenza è confermata da casi di schizencefalia in fratelli e sorelle.
Lo sviluppo della malattia può essere influenzato infezioni (ad esempio, citomegalovirus) e medicinali .
Quali processi provocano la comparsa dello spazio nella materia grigia?
Altri hanno un'opinione diversa: di conseguenza si formano delle fessure nella materia grigia occlusione vascolare . Il blocco o l'assenza della carotide interna o delle arterie cerebrali medie porta a ictus ischemico e successivamente a necrosi cerebrale.
Diagnostica
L'esame e il trattamento sintomatico vengono effettuati nel reparto neuropsichiatrico.I medici utilizzano i seguenti metodi diagnostici strumentali:
- Risonanza magnetica.
- Tomografia computerizzata a raggi X.
- L'elettroencefalografia è integrata da test con apertura e chiusura degli occhi, fotostimolazione e iperventilazione (al bambino viene chiesto di inspirare ed espirare rapidamente e profondamente).
In tutti i bambini affetti da schizencefalia, l'EEG mostra un rallentamento dell'attività di fondo, nonché uno dei due cambiamenti:
- attività epilettica locale nelle regioni frontotemporali;
- attività epilettica diffusa senza un focus specifico.
A causa della presenza dell'idrocefalo, la schizencefalia aperta è simile a porencefalia , tuttavia, nel secondo caso, la fessura è ricoperta non da tessuto epiteliale, ma da tessuto connettivo o gliale (ausiliario). La malattia può essere confusa con oloprosencefalia (assenza completa o parziale di divisione del proencefalo in emisferi).
La TC viene utilizzata raramente nella diagnosi della schizencefalia, poiché la risonanza magnetica fornisce un quadro più completo della patologia.

La risonanza magnetica può rilevare disturbi concomitanti dello sviluppo cerebrale:
- eterotopia della materia grigia (noduli nella materia grigia sotto il rivestimento dei ventricoli);
- ipoplasia del nervo ottico (numero insufficiente di assoni, unità strutturali di neuroni);
- agenesia del setto pellucida con localizzazione frontale della schizencefalia;
- displasia setto-ottica (disturbi dello sviluppo della ghiandola pituitaria, del setto pellucido, del nervo ottico).
Trattamento
Viene fornito il trattamento sintomatico della schizencefalia.
Tetraparesi, emiparesi, convulsioni, spasticità muscolare, ritardo dello sviluppo psicomotorio, vengono trattate con stimolazione elettrica o micropolarizzazione del cervello, psicoterapia, farmaci antiepilettici, viene utilizzata la terapia botulinica (bloccando la trasmissione di segnali indesiderati dai nervi ai muscoli), viene utilizzato il trattamento ortopedico .
I pazienti con schizencefalia lieve non presentano ricadute dopo aver iniziato il trattamento con farmaci antiepilettici.
Quali medici oltre al neurologo e al neurochirurgo aiuteranno il bambino?
Medici di almeno 3 specialità possono aiutare a migliorare la qualità della vita:
- Fisioterapista prescriverà una terapia per migliorare la prognosi dello sviluppo motorio, vale a dire la capacità di sedersi e stare in piedi (nei casi più gravi). I bambini con sintomi moderati possono trarre beneficio da esercizi per rafforzare i muscoli delle braccia e delle gambe.
- Servizi terapista occupazionale sarà necessario se il bambino non può eseguire azioni che richiedono capacità motorie fini ben sviluppate: mangiare, vestirsi in modo indipendente. La terapia occupazionale consentirà di vivere una vita piena e di svolgere funzioni a casa, all'asilo e a scuola.
- Logopedista Migliora le capacità di parlare e deglutire.
Qual è la prognosi?
La schizencefalia ha una prognosi prevalentemente favorevole per la vita. In caso di fornitura tempestiva di misure di rianimazione e/o riabilitazione e successivo trattamento, si verifica la remissione. I problemi con l'attività motoria persisteranno per tutta la vita e esiste il rischio di ritardo mentale, ma la maggior parte dei pazienti può vivere pienamente nella società.Oltre all'epilessia, il problema principale dei pazienti affetti da schizencefalia è l'idrocefalo. Con un costante aumento del fluido da un lato, i ventricoli vengono spostati e i tessuti circostanti vengono compressi, compreso il midollo allungato (regola l'attività del cuore e la funzione respiratoria). L’idrocefalo moderato viene trattato farmacologicamente, ma i medici non sempre offrono opzioni diverse dallo shunt.
Storia di un piccolo paziente: ragazzo, 2 anni.
Madre - 25 anni, padre - 29 anni, prima gravidanza, salute soddisfacente, assenza di fattori ambientali dannosi nella zona di residenza e sul lavoro.
L'idrocefalo è stato suggerito per la prima volta dall'ecografia a 34 settimane. Dalla clinica distrettuale la paziente è stata indirizzata al centro perinatale regionale.
La dimensione del feto alla fetometria corrispondeva all'età gestazionale. Durante l'esame del cervello, è stata notata una cavità con contenuto liquido nell'emisfero destro. I glomeruli vascolari in esso contenuti hanno permesso di verificare che la causa della sua formazione non era una cisti. A parte il cerchio aperto di Willis, non sono stati rilevati altri cambiamenti.
È stata fatta una diagnosi clinica: schizencefalia di tipo 2 (con schisi aperta). Dopo 5 settimane nacque un bambino maschio. Peso: 3450 g, 7 punti della scala Apgar. Immediatamente dopo la nascita è stata eseguita una NSG e la diagnosi è stata confermata. Madre e bambino sono stati dimessi dall'ospedale di maternità il 4° giorno.
sono passati 2 anni. Il bambino è molto indietro rispetto ai suoi coetanei nello sviluppo psicomotorio (statica, capacità motorie, reazioni sensoriali, linguaggio, interazione sociale), le capacità motorie sono limitate. Si osservano sindrome convulsiva e diminuzione dei riflessi spinali.
La presenza di anomalie craniofacciali visibili visivamente ha un significato prognostico negativo: microcefalia, forma della testa idrocefalica. Deviazioni simili possono svilupparsi in un bambino con schizencefalia aperta.
Un bambino con schizencefalia chiusa riceverà una prognosi favorevole per la vita. Le fessure aperte nella materia grigia, al contrario, portano a un ritardo dello sviluppo mentale o psico-linguistico (ZPR o ZPRR) e a disturbi del movimento.
Anamnesi di un paziente adulto: 20 anni.
Gestione dei disturbi del torcicollo (rumore e ronzio nelle orecchie), crisi epilettiche con automatismi del linguaggio (pronuncia incontrollata delle parole), crisi tonico-cloniche. Gli attacchi di epilessia portano alla perdita di coscienza.
Dal momento della nascita fino alla visita in ospedale dopo l'ultima crisi avvenuta durante le lezioni all'università, non ci si aspettava la diagnosi di schizencefalia.
Breve anamnesi. Alla nascita non sono state notate deviazioni; il ritardo dello sviluppo è iniziato a 9 mesi; la parte destra ha improvvisamente smesso di obbedire. Dopo aver contattato un neurologo pediatrico, hanno eseguito una risonanza magnetica e una TC e hanno diagnosticato una paralisi cerebrale (in seguito si è scoperto che la diagnosi non era corretta). È stato prescritto un ciclo di farmaci vasoattivi e neurometabolici, sebbene non vi fossero indicazioni corrispondenti.
Il primo attacco di epilessia si verificò all’età di 8 anni. Successivamente sono stati osservati attacchi con aura uditiva e gravi convulsioni, ma senza perdita di coscienza. Furono prescritti molti farmaci, compresi gli antiepilettici, ma la malattia progredì.
Recentemente, gli attacchi sono iniziati pochi giorni prima o all'inizio delle mestruazioni. Per il trattamento dell'epilessia è stato prescritto un ciclo di Depakine in combinazione con Lamictal. Il numero di crisi è diminuito, ma quando sono iniziate si sono verificate diverse crisi al giorno.
Risultati diagnostici quando si visita un ospedale clinico regionale. L'EEG ha registrato cambiamenti moderati nell'attività bioelettrica, nel ritmo alfa irregolare e nell'epiattività nella regione temporale dell'emisfero sinistro. L'immagine MRI è caratteristica della schizencefalia.
Difetti estetici: strabismo divergente, asimmetria della zona nasolabiale, palato gotico (alto e stretto, arcuato), la forma delle arcate dentarie è disturbata, ittiosi (pelle secca e squamosa) nella parte inferiore delle gambe, il braccio e la gamba destra sono accorciati di 2 e 2,5 cm.
Problemi neurologici: astigmatismo (parziale offuscamento dei contorni dell'immagine, visione offuscata), sul lato destro del corpo si osserva un aumento dei riflessi tendinei (crampi muscolari in allungamento), paraparesi (diminuzione dell'attività muscolare), diminuzione della sensibilità. Instabile nella posizione di Romberg (in piedi con le braccia tese). Polineuropatia (ridotta sensibilità nelle braccia sotto il gomito, ipersensibilità nelle gambe sotto il ginocchio).
Una prognosi pessimistica viene data ai bambini con epilessia resistente ai farmaci (cioè con convulsioni che non possono essere controllate con i farmaci). La presenza di patologie concomitanti peggiora la qualità della vita e riduce le opportunità disponibili.
Un esito letale è possibile in caso di infezioni acute (comprese quelle diventate croniche), disordini metabolici, tossicosi grave e insufficienza multiorgano.
Eterotopia subependimale(eterotopia periventricolare) è la forma più comune di eterotopia della sostanza grigia (GM), caratterizzata da noduli SV localizzati direttamente sotto l'ependima dei ventricoli laterali. In base alla morfologia si può dividere in:
- focale unilaterale
- focale bilaterale
- diffusa bilaterale: una striscia ondulata di SV che circonda i ventricoli.
Epidemiologia
La maggior parte dei casi sono sporadici, alcuni sono recessivi legati all'X (Xq28). Le donne sperimentano un deterioramento cognitivo relativamente lieve e successivamente sviluppano l'epilessia. Nel caso dei maschi si osserva l'interruzione spontanea della gravidanza, solitamente a causa di malformazioni del sistema cardiovascolare. I sopravvissuti hanno gravi disabilità.
Quadro clinico
Molto spesso, l'eterotopia subependimale è associata a epilessia e ritardo dello sviluppo.
Patologia
Come altri tipi di eterotopie, questo tipo è il risultato di una ridotta migrazione neuronale. In alcuni casi, la causa dello sviluppo dell'eterotopia subependimale è una violazione della proliferazione cellulare.
I noduli di materia grigia sono costituiti da gruppi di neuroni e cellule gliali. È interessante notare che sono più comuni sul lato destro, presumibilmente a causa della successiva migrazione dei neuroblasti sul lato destro.
Nei casi legati all'X, si osservano mutazioni nel gene per la filamina-1, una proteina che reticola l'actina intracellulare. Inoltre, la filamina-1 svolge anche un ruolo importante nello sviluppo vascolare.
Diagnostica
La RM è il metodo di scelta, sebbene l'eterotopia periventricolare sia visibile alla TC e all'ecografia (se molto grande).
Ultrasuoni
I noduli SV subependimali sono solitamente iperecogeni rispetto alla sostanza bianca normale e possono sporgere nel lume ventricolare (il bordo ondulato del ventricolo).
CT
Alla TC, l'eterotopia subependimale appare come un'area di tessuto non calcificata che non accumula materiale di contrasto, simile per densità alla normale materia grigia, attorno ai ventricoli laterali.
risonanza magnetica
RM prenatale
Alla fine della gravidanza, la diagnosi di eterotopia subependimale è relativamente ovvia. Prima della 26a settimana di gravidanza, la presenza di una normale matrice germinale periventricolare telencefalica ne rende difficile l'individuazione, così come il movimento fetale.
RM postnatale
Nello strato ependimale si osservano piccoli noduli di sostanza grigia che distorcono il contorno dei ventricoli. Molto spesso localizzato nella regione del triangolo e delle corna occipitali. Altre parti del cervello sembrano normali.
I noduli di materia grigia vengono visualizzati in tutte le sequenze, comprese quelle post-contrasto, dove, come la normale materia grigia, non accumulano materiale di contrasto.
Diagnosi differenziale
- norma
- nuclei caudati
- talamo
- Astrocitoma subependimale a cellule giganti
- ha un pronunciato accumulo di contrasto
- localizzato vicino al forame di Monroe
- linfonodi subependimali nella sclerosi tuberosa
- solitamente calcificato (tranne nella prima infanzia)
- segnale T2 più alto rispetto al segnale della materia grigia
- emorragia subependiale all'ecografia e alla risonanza magnetica prenatale
- sebbene il quadro possa essere simile, uno studio di controllo in caso di emorragia determina l'evoluzione dei cambiamenti
Questo è il risultato di disturbi nella formazione delle singole strutture cerebrali o del cervello nel suo insieme che si verificano nel periodo prenatale. Spesso presentano sintomi clinici aspecifici: sindrome prevalentemente epilettica, ritardo mentale e mentale. La gravità del quadro clinico è direttamente correlata al grado di danno cerebrale. Vengono diagnosticati prima della nascita durante l'ecografia ostetrica, dopo la nascita, utilizzando l'EEG, la neurosonografia e la risonanza magnetica del cervello. Il trattamento è sintomatico: antiepilettico, disidratazione, metabolico, psicocorrettivo.
ICD-10
Q00 Q01 Q02 Q04

informazioni generali
Le anomalie dello sviluppo cerebrale sono difetti costituiti da cambiamenti anormali nella struttura anatomica delle strutture cerebrali. La gravità dei sintomi neurologici che accompagnano le anomalie cerebrali varia in modo significativo. Nei casi più gravi, i difetti sono la causa della morte prenatale del feto; rappresentano fino al 75% dei casi di morte intrauterina. Inoltre, gravi anomalie cerebrali rappresentano circa il 40% delle morti neonatali. I tempi di manifestazione dei sintomi clinici possono variare. Nella maggior parte dei casi, le anomalie cerebrali compaiono nei primi mesi dopo la nascita del bambino. Ma, poiché la formazione del cervello dura fino all'età di 8 anni, una serie di difetti fanno il loro debutto clinico dopo il 1° anno di vita. In più della metà dei casi, i difetti cerebrali sono associati a difetti degli organi somatici. L'individuazione prenatale delle anomalie cerebrali è un compito urgente nella pratica ginecologica e ostetrica, e la loro diagnosi e trattamento postnatale sono questioni prioritarie della moderna neurologia, neonatologia, pediatria e neurochirurgia.
Cause
La causa più significativa di interruzioni nello sviluppo intrauterino è l'influenza sul corpo della donna incinta e sul feto di vari fattori dannosi che hanno un effetto teratogeno. Il verificarsi di un'anomalia derivante dall'eredità monogenica si verifica solo nell'1% dei casi. La causa più influente dei difetti cerebrali è considerata un fattore esogeno. Molti composti chimici attivi, contaminazione radioattiva e alcuni fattori biologici hanno un effetto teratogeno. Di non poca importanza qui è il problema dell'inquinamento dell'ambiente umano, che provoca l'ingresso di sostanze chimiche tossiche nel corpo di una donna incinta.
Vari effetti embriotossici possono essere associati allo stile di vita della donna incinta stessa: ad esempio fumo, alcolismo, tossicodipendenza. Anche i disturbi dismetabolici in una donna incinta, come il diabete mellito, l'ipertiroidismo, ecc., possono causare anomalie cerebrali nel feto. Molti farmaci che una donna può assumere nelle prime fasi della gravidanza, ignara dei processi che si verificano nel suo corpo, hanno anche un effetto teratogeno. Le infezioni subite da una donna incinta o le infezioni intrauterine del feto hanno un potente effetto teratogeno. I più pericolosi sono la citomegalia, la listeriosi, la rosolia e la toxoplasmosi.
Patogenesi
La costruzione del sistema nervoso fetale inizia letteralmente dalla prima settimana di gravidanza. Già dal 23 ° giorno di gestazione termina la formazione del tubo neurale, la cui fusione incompleta dell'estremità anteriore comporta gravi anomalie cerebrali. Intorno al 28 ° giorno di gravidanza si forma la vescicola cerebrale anteriore, che successivamente si divide in 2 laterali, che costituiscono la base degli emisferi cerebrali. Successivamente si formano la corteccia cerebrale, le sue convoluzioni, il corpo calloso, le strutture basali, ecc.
La differenziazione dei neuroblasti (cellule nervose germinali) porta alla formazione di neuroni, che formano la materia grigia, e di cellule gliali, che costituiscono la materia bianca. La materia grigia è responsabile dei processi più elevati dell'attività nervosa. La sostanza bianca contiene vari percorsi che collegano le strutture cerebrali in un unico meccanismo funzionante. Un neonato a termine ha lo stesso numero di neuroni di un adulto. Ma lo sviluppo del suo cervello continua, soprattutto intensamente nei primi 3 mesi. vita. C'è un aumento delle cellule gliali, della ramificazione dei processi neuronali e della loro mielinizzazione.
I fallimenti possono verificarsi in vari stadi della formazione del cervello. Se si verificano nei primi 6 mesi. gravidanza, possono portare ad una diminuzione del numero di neuroni formati, a vari disturbi nella differenziazione e all'ipoplasia di varie parti del cervello. In un secondo momento possono verificarsi danni e morte della sostanza cerebrale normalmente formata.
Tipi di anomalie cerebrali
Anencefalia- assenza di cervello e acrania (assenza di ossa del cranio). Il posto del cervello è occupato da escrescenze di tessuto connettivo e cavità cistiche. Può essere coperto di pelle o nudo. La patologia è incompatibile con la vita.
Encefalocele- prolasso dei tessuti e delle membrane cerebrali attraverso un difetto delle ossa del cranio, causato dalla mancata fusione. Di norma, si forma lungo la linea mediana, ma può anche essere asimmetrico. Un piccolo encefalocele può simulare un cefaloematoma. In questi casi, la radiografia del cranio aiuta a determinare la diagnosi. La prognosi dipende dalle dimensioni e dal contenuto dell'encefalocele. Se la protrusione è di piccole dimensioni e nella sua cavità è presente tessuto nervoso ectopico, la rimozione chirurgica dell'encefalocele è efficace.
Microcefalia- riduzione del volume e del peso del cervello a causa di un ritardo nel suo sviluppo. Si verifica con una frequenza di 1 caso ogni 5mila neonati. Accompagnato da una circonferenza cranica ridotta e da un rapporto sproporzionato cranio facciale/cranico con predominanza del primo. La microcefalia rappresenta circa l'11% di tutti i casi di ritardo mentale. Con grave microcefalia, l'idiozia è possibile. Spesso non c'è solo ritardo mentale, ma anche un ritardo nello sviluppo fisico.
Macrocefalia- aumento del volume e della massa cerebrale. Molto meno comune della microcefalia. La macrocefalia è solitamente associata a disturbi dell'architettura cerebrale e all'eterotopia focale della sostanza bianca. La principale manifestazione clinica è il ritardo mentale. Può verificarsi una sindrome convulsiva. La macrocefalia parziale si verifica con l'allargamento di solo uno degli emisferi. Di norma, è accompagnato dall'asimmetria della parte cerebrale del cranio.
Displasia cerebrale cistica- caratterizzato da molteplici cavità cistiche del cervello, solitamente collegate al sistema ventricolare. Le cisti possono variare di dimensioni. A volte sono localizzati solo in un emisfero. Cisti cerebrali multiple si presentano con epilessia resistente alla terapia anticonvulsivante. Le singole cisti, a seconda delle loro dimensioni, possono avere un decorso subclinico o essere accompagnate da ipertensione endocranica; il loro graduale riassorbimento è spesso annotato.
Oloprosencefalia- assenza di separazione degli emisferi, per cui sono rappresentati da un unico emisfero. I ventricoli laterali sono formati in un'unica cavità. Accompagnato da grave displasia del cranio facciale e difetti somatici. La natimortalità o la morte si verificano il primo giorno.
Displasia corticale focale(FCD) - la presenza nella corteccia cerebrale di aree patologiche con neuroni giganti e astrociti anormali. La posizione preferita sono le aree temporali e frontali del cervello. Una caratteristica distintiva delle crisi epilettiche durante la FCD è la presenza di parossismi complessi a breve termine con rapida generalizzazione, accompagnati nella loro fase iniziale da fenomeni motori dimostrativi sotto forma di gesti, calpestio in un punto, ecc.
Eterotopie- accumuli di neuroni che, nella fase di migrazione neuronale, erano ritardati nel loro percorso verso la corteccia. Le eterotopioni possono essere singole o multiple, avere una forma nodulare o nastro. La loro principale differenza rispetto alla sclerosi tuberosa è la mancanza della capacità di accumulare contrasto. Queste anomalie dello sviluppo cerebrale si manifestano con episindrome e ritardo mentale, la cui gravità è direttamente correlata al numero e alla dimensione degli eterotopioni. Con l'eterotopia singola, le crisi epilettiche, di regola, debuttano dopo i 10 anni di età.
Diagnostica
Gravi anomalie cerebrali possono spesso essere diagnosticate mediante esame visivo. In altri casi, l'anomalia cerebrale può essere sospettata da ritardo cerebrale, ipotonia muscolare nel periodo neonatale e comparsa di sindrome convulsiva nei bambini nel primo anno di vita. La natura traumatica o ipossica del danno cerebrale può essere esclusa se non vi è alcuna storia di trauma alla nascita del neonato, ipossia fetale o asfissia del neonato. La diagnosi prenatale delle malformazioni fetali viene effettuata mediante screening ecografico durante la gravidanza. L'ecografia nel primo trimestre di gravidanza può prevenire la nascita di un bambino con grave anomalia cerebrale.
Uno dei metodi per identificare i difetti cerebrali nei neonati è la neurosonografia attraverso la fontanella. Dati molto più accurati nei bambini di qualsiasi età e negli adulti si ottengono utilizzando la risonanza magnetica del cervello. La risonanza magnetica consente di determinare la natura e la localizzazione dell'anomalia, la dimensione delle cisti, delle eterotopie e di altre aree anomale e di effettuare una diagnosi differenziale con lesioni cerebrali ipossiche, traumatiche, tumorali e infettive. La diagnosi della sindrome convulsiva e la selezione della terapia anticonvulsivante vengono effettuate utilizzando l'EEG, nonché il monitoraggio video EEG prolungato. Se ci sono casi familiari di anomalie cerebrali, può essere utile la consultazione di un genetista con ricerche genealogiche e analisi del DNA. Per identificare anomalie combinate, viene effettuato l'esame degli organi somatici: ecografia del cuore, ecografia della cavità addominale, radiografia degli organi del torace, ecografia dei reni, ecc.
Trattamento delle anomalie cerebrali
Il trattamento delle malformazioni cerebrali è prevalentemente sintomatico, effettuato da un neurologo pediatrico, neonatologo, pediatra ed epilettologo. In presenza di sindrome convulsiva viene effettuata la terapia anticonvulsivante (carbamazepina, levetiracetam, valproato, nitrazepam, lamotrigina, ecc.). Poiché l'epilessia nei bambini, che accompagna anomalie dello sviluppo cerebrale, è solitamente resistente alla monoterapia anticonvulsivante, viene prescritta una combinazione di 2 farmaci (ad esempio levetiracetam con lamotrigina). Per l'idrocefalo viene eseguita la terapia di disidratazione e le operazioni di shunt vengono utilizzate secondo le indicazioni. Al fine di migliorare il metabolismo del tessuto cerebrale normalmente funzionante, compensando in una certa misura il difetto congenito esistente, è possibile condurre un ciclo di trattamento neurometabolico con la somministrazione di glicina e vitamine. B, ecc. I farmaci nootropici vengono utilizzati nel trattamento solo in assenza di episindrome.
Per anomalie cerebrali moderate e relativamente lievi, si raccomanda un supporto psicologico completo per il bambino e l'educazione dei bambini più grandi in scuole specializzate. Queste tecniche aiutano a instillare capacità di cura di sé, a ridurre la gravità del ritardo mentale e, se possibile, ad adattare socialmente i bambini con difetti cerebrali.
Prognosi e prevenzione
La prognosi è in gran parte determinata dalla gravità dell'anomalia cerebrale. Un sintomo sfavorevole è l'insorgenza precoce dell'epilessia e la sua resistenza alla terapia. La prognosi è complicata dalla presenza di concomitante patologia somatica congenita. Una misura preventiva efficace è l'esclusione degli effetti embriotossici e teratogeni su una donna durante la gravidanza. Quando pianificano una gravidanza, i futuri genitori dovrebbero liberarsi delle cattive abitudini, sottoporsi a consulenza genetica ed essere sottoposti a screening per le infezioni croniche.

Principali regioni morfologiche del cervello
- Il cervello proencefalo (estremità) è costituito da due emisferi cerebrali.
- Il diencefalo è costituito da talamo, epitalamo, ipotalamo e ghiandola pituitaria, che non è inclusa nel diencefalo, ma è separata in una ghiandola separata.
- Il mesencefalo è costituito dai peduncoli cerebrali e dal tetto della regione quadrigeminale. I collicoli superiori del tetto quadrigemino sono il centro visivo sottocorticale, mentre i collicoli inferiori sono il centro uditivo sottocorticale.
- Il rombencefalo è costituito dal ponte e dal cervelletto.
- midollo. La giunzione del midollo allungato e del midollo spinale è il forame magno.
Il mesencefalo, il rombencefalo e il midollo allungato sono combinati nel tronco encefalico.

Struttura interna degli emisferi cerebrali.
- materia grigia
- materia bianca
La materia grigia è costituita dalla corteccia, che ricopre completamente gli emisferi cerebrali. La materia bianca si trova sotto la materia grigia del cervello. Tuttavia, la sostanza bianca contiene anche aree di materia grigia, ovvero gruppi di cellule nervose. Si chiamano nuclei. Normalmente esiste un confine netto tra la materia bianca e quella grigia. La differenziazione della sostanza bianca e grigia è possibile alla TC, ma è meglio differenziata alla RM.

Displasia corticale
Con la displasia corticale, i confini tra la materia bianca e quella grigia sono sfumati. In questo caso, dovrebbe essere utilizzata anche la sequenza di recupero dell'inversione T1. Su queste immagini saranno visibili i bordi, ad eccezione delle aree di displasia corticale.

Attacco di cuore
Nell'edema citotossico, che si sviluppa nei primi minuti di infarto cerebrale, si perde anche la differenziazione tra materia bianca e grigia, che è un segno TC precoce di infarto cerebrale.







Emisferi maggiori del cervello
Gli emisferi del cervello sono separati l'uno dall'altro da un ampio processo falciforme. Ogni emisfero ha 4 lobi:
- Lobo frontale.
- Lobo parietale
- Lobo occipitale
Il lobo frontale viene separato dal lobo parietale mediante il solco centrale o ralandico, chiaramente visibile sia nella sezione assiale che in quella sagittale.
Il lobo frontale è separato dal lobo temporale mediante il solco laterale, che è chiaramente visualizzato sia in sezione sagittale che assiale, nonché nelle sezioni frontali.
Il lobo parietale è separato dal lobo occipitale dall'omonimo solco parieto-occipitale. Questa linea separa anche i bacini carotideo e basilare.
Alcuni autori identificano l'insula in un solco separato, che è una vasta area della corteccia, che copre l'insula superiormente e lateralmente, forma un opercolo (lat. pars opercularis) ed è formato da parte dell'adiacente frontale, temporale e parietale lobi.
Condividere i confini


Condividere i confini
Confini dei lobi frontali e parietali.
Omega-?
Solco centrale
Sintomo dei baffi– giro postcentrale.
Giro del cingolo – giro postcentrale.
Per determinare correttamente il confine dei lobi frontali e parietali, troviamo innanzitutto il solco centrale. Il simbolo si inserisce in questa scanalatura Omega –? su sezioni assiali.
Aiutano anche il sintomo di un baffo situato perpendicolare alla linea mediana e l'immagine, che corrisponde al solco postcentrale. Anteriormente al giro postcentrale si trova il solco centrale.
Il solco cingolato.
Nelle sezioni sagittali occorre individuare il corpo calloso; al di sopra di esso è presente un solco cingolato, che prosegue posteriormente e verso l'alto nel solco postcentrale, da cui anteriormente si localizza il solco centrale o Rolandico.

Lobo frontale
Il lobo frontale è grande e uno dei giri principali è il giro precentrale, che è il centro corticale del movimento. Il lobo frontale contiene anche il giro superiore, medio e inferiore. Le convoluzioni elencate vanno dall'alto verso il basso e sono parallele tra loro.
Sulla superficie inferiore del lobo frontale sono presenti giri retti e orbitali, tra i quali si trovano i tratti olfattivi e i bulbi. Queste aree sono danneggiate da lesioni.

Lesione traumatica al lobo frontale
In questo paziente notiamo un danno simmetrico alle parti basali di entrambi i lobi frontali, che corrispondono a cambiamenti post-traumatici.

zona di Broca
Un'altra area importante è l'area di Broca, che si trova nelle parti distali del giro frontale inferiore. La sua localizzazione è importante quando si pianificano interventi neurochirurgici. Questa zona è facile da trovare, ricordando l'icona di McDonald's.

Infarto che coinvolge l'area di Broca nel processo patologico
Questo paziente ha un infarto acuto causato dall'occlusione del ramo anteriore della M2 della MCA sinistra. Danno al lobo frontale che coinvolge l'area di Broca nel processo patologico.

Lobo parietale
Dietro il solco centrale si trova il giro postcentrale, che funge da analizzatore corticale della sensibilità generale e propriocettiva.
Posteriormente si trovano i lobuli parietali superiore ed inferiore.
Nel lobulo parietale superiore si trova il nucleo dell'analizzatore cutaneo, responsabile della stereognosi, la capacità di riconoscere gli oggetti al tatto.
Nel lobulo parietale inferiore è presente un analizzatore motorio responsabile dell'aprassia: movimenti intenzionali e volontari.
Stereognosia- la capacità di riconoscere gli oggetti al tatto.
Aprassia- violazione di azioni volontarie.

Atrofia del precuneo
L'atrofia del precuneo è un sintomo precoce della malattia di Alzheimer, ancor prima dell'atrofia della corteccia del lobo temporale e dell'ippocampo.
Il precuneo è una regione del lobo parietale sulla superficie interna di entrambi gli emisferi del cervello, situata sopra e davanti al corpo calloso.

Lobo temporale
Nel lobo temporale c'è
Giro temporale superiore
Giro temporale medio
Giro temporale inferiore. Queste tre circonvoluzioni sono parallele tra loro e situate su un piano orizzontale.
Le circonvoluzioni di Heschl si trovano sulla superficie del giro temporale superiore. Sono il centro corticale dell'udito.
Il giro paraippocampale si trova sulla superficie inferiore dei lobi temporali nelle sezioni mediali. L'uncino, insieme all'ippocampo, è responsabile dell'olfatto. Quando l’ippocampo è danneggiato, la memoria è la prima ad essere colpita.
La zona di Wernicke. L'area di Wernicke si trova nelle parti distali della circonvoluzione temporale superiore. È un'area sensoriale del linguaggio.

Lobo occipitale
Nei lobi occipitali si definiscono solchi e convoluzioni non permanenti, ma il più costante è il solco calcarino, situato sulla superficie mediale del lobo occipitale. Intorno al solco calcarino si trovano le aree di Brodmann 17, 18 e 19, che sono il centro corticale della visione.

Occlusione della PCA
Questo paziente presenta clinicamente un deficit visivo causato da un danno al lobo occipitale causato da un infarto (occlusione della PCA).
Materia grigia sottocorticale







Materia grigia sottocorticale
La materia grigia sottocorticale comprende:
- talamo
- gangli della base
- nucleo caudato
- nucleo lenticolare, che contiene il putamen e il globo pallido.
- conchiglia
La capsula interna è costituita dalla parte anteriore della coscia, dal ginocchio e dalla parte posteriore della coscia.
Come trovare l'anca posteriore?
Tra il talamo ed il nucleo lenticolare troviamo un fuoco iperintenso, che è il tratto piramidale. Da questo fuoco iperintenso tracciamo una linea fino al ginocchio, che sarà la proiezione della parte posteriore della coscia della capsula interna.
NB – non confondere il ginocchio posteriore con il globo pallido.
Quando si classificano le emorragie intracerebrali nella sostanza grigia sottocorticale, a seconda della posizione rispetto alla capsula interna, le emorragie si dividono in:
- laterale
- mediale
- misto
SOSTANZA BIANCA
Fibre commissurali che collegano tra loro gli emisferi.
Corpo calloso (commissura più grande)
Commissura anteriore
Commissura posteriore (commissura del fornice)

Commissura anteriore
La commissura anteriore si trova sotto il becco del corpo calloso dietro la placca terminale e collega alcune parti del cervello olfattivo: il giro ippocampale, gli uncusi sinistro e destro dei lobi temporali.
Commissura posteriore
La commessura posteriore appartiene all'epitalamo, si trova alla radice dell'epifisi e collega le parti corrispondenti del mesencefalo e del diencefalo.
Significato pratico:
Per valutare il corpo calloso viene utilizzata una linea bicommissurale sul piano sagittale. La linea bicommissurale viene tracciata attraverso il bordo superiore della commissura anteriore e il bordo inferiore della commissura posteriore.

corpo calloso
Il corpo calloso è costituito da:
Tronco o corpo (anteriore e posteriore)
Ogni sezione collega la parte omolaterale del cervello.
Formazione del corpo calloso.
Il corpo calloso si sviluppa in un ordine speciale:
Dal ginocchio si sviluppa poi il corpo, la cresta ed infine il becco.
La mielinizzazione del corpo calloso procede dalle sezioni posteriori alle sezioni anteriori.
Questa conoscenza aiuta a restringere la diagnosi differenziale per le patologie del corpo calloso.

Disgenesia e atrofia del corpo calloso
Nella disgenesia del corpo calloso, il ginocchio e le parti anteriori del corpo calloso sono ben formati, ma lo splenio e il becco sono assenti. Questa patologia è congenita. La patologia è presentata a sinistra.
Con l'atrofia del corpo calloso, le parti posteriori del corpo calloso sono ben formate (la parte posteriore del corpo e lo splenio), ma il becco, il ginocchio e la parte anteriore del corpo sono di dimensioni ridotte. Queste modifiche vengono acquistate.
Molte malattie colpiscono il corpo calloso, quindi la presenza di lesioni non è patognomonica per una particolare malattia.

Malattia di Marchiafava-Bignami
Malattia di Marchiafava-Bignami (degenerazione centrale del corpo calloso, sindrome di Marchiafava, mielinolisi extrapontina).
Si verifica nelle persone che abusano di alcol. In questi individui, la risonanza magnetica rivela lesioni dello splenio e delle parti posteriori del tronco (corpo) del corpo calloso.
Negli stadi cronici della malattia di Marchiafava-Bignami, il corpo calloso si presenta sotto forma di sandwich, in cui sono conservati gli strati superiore ed inferiore del corpo calloso, ma con necrosi degli strati intermedi.
materia bianca
Materia bianca:
- periventricolare
- sezioni profonde (centri semiovali)
- Fibra a U
La sostanza bianca periventricolare si trova in prossimità dei ventricoli laterali del cervello.
Le fibre a U collegano la corteccia dei giri vicini o della sostanza bianca sottocorticale.
Sostanza bianca profonda situata tra la sostanza bianca periventricolare e sottocorticale.
Lesioni della sostanza bianca:
Le lesioni della sostanza bianca sono classificate in base alla localizzazione:
- periventricolare
- giustacorticale
- sottocorticale
- lesioni nella sostanza bianca profonda

Lesioni periventricolari
periventricolare (singolo o multiplo, piccolo o grande, che si fonde tra loro)

Lesioni iuxtacorticali
juxta: circa. Queste lesioni sono localizzate nelle fibre U e sono direttamente adiacenti alla sostanza grigia, cioè non c'è nessuno strato di sostanza bianca tra la lesione e la sostanza grigia.
Queste lesioni possono avere forma diversa, come ripetere la forma delle fibre a U, ma possono anche essere di forma rotonda o irregolare. Questa localizzazione è patognomonica per la SM.

Lesioni sottocorticali
Le lesioni sottocorticali sono lesioni localizzate vicino alla corteccia cerebrale, ma tra la lesione e la corteccia è presente uno strato di sostanza bianca.

Lesioni nella sostanza bianca profonda.
Queste lesioni si verificano in varie malattie del cervello.




VENTRICOLI DEL CERVELLO
I ventricoli laterali sono costituiti da:
- corna anteriori (frontali).
- corna posteriori (occipitali).
- corna inferiori (temporali).
I ventricoli laterali sono collegati al terzo ventricolo tramite i forami accoppiati di Monroe.
Il terzo ventricolo ha forma irregolare per la presenza di tasche. L'apertura del terzo ventricolo corrisponde alla commissura intertalamica.
Il terzo ventricolo è collegato al quarto ventricolo tramite l'acquedotto Silviano. Dal quarto ventricolo, il liquido cerebrospinale entra nelle cisterne basali attraverso i forami accoppiati di Luschka e l'apertura spaiata di Maugendie.
Quando si valutano i ventricoli, vale la pena prestare attenzione ai corni ventricolari, poiché nelle malattie degenerative come il morbo di Alzheimer, l'atrofia dell'ippocampo è accompagnata dall'espansione delle corna temporali. Nella modalità FLAIR aumenta il segnale proveniente dalle corna posteriori (occipitale), che è la norma, così come l'asimmetria delle corna.
TERZO VENTRICOLO.
Il terzo ventricolo si trova sulla linea mediana tra il talamo ottico. È collegato ai ventricoli laterali tramite il forame di Monroe e al quarto ventricolo tramite l'acquedotto cerebrale.
Tasche del terzo ventricolo:
- Soprachiasmatico
- Infundibolare
- Soprapineale
- Pineale
Normalmente, queste tasche hanno angoli acuti, ma quando la pressione aumenta, le tasche si aprono.
Quarto ventricolo del cervello.
Il quarto ventricolo è la cavità del rombencefalo e, attraverso i fori pari di Luschka e i fori spaiati di Magendie, è collegato alle cisterne basali.

Plessi coroidei
I plessi corioidei che producono liquido cerebrospinale si trovano in tutti i ventricoli del cervello, quindi la calcificazione del plesso coroideo, che è più spesso visualizzata nei corni dorsali dei ventricoli laterali, può essere vista sia nel terzo che nel quarto ventricolo.

Sclerosi tuberosa.
La calcificazione del plesso coroideo, che è normale, non deve essere confusa con condizioni patologiche. Ad esempio, con calcificazioni dei ventricoli laterali - tuberi periventricolari nella sclerosi tuberosa.

Eterotopia della materia grigia
È importante ricordare che l'unica materia grigia che confina con i ventricoli laterali sono i nuclei caudati, che hanno contorni chiari e uniformi. Ulteriori strutture della materia grigia che deformano il contorno dei ventricoli laterali sono cambiamenti patologici caratteristici dell'eterotopia della materia grigia.

Varianti della struttura dei ventricoli
- la cavità del setto trasparente, che si osserva nella maggior parte dei neonati (si chiude nel tempo) e sembra una forma triangolare tra i corpi del ventricolo laterale anteriore. Questa cavità non attraversa mai il forame di Monroe.
- cavità velica intermedia. Una delle pareti della cavità, che costituisce il tetto del terzo ventricolo.
- La cavità di Verge è una cavità estesa tra i corpi dei ventricoli laterali.

Cisti colloide
È necessario distinguere le varianti strutturali della cisti colloidale, che differiranno dall'intensità del segnale proveniente dal liquido cerebrospinale in quasi tutte le sequenze di impulsi. Dopo la somministrazione di un mezzo di contrasto, le cisti colloidali non accumulano contrasto, il che corrisponde ad un processo benigno.

MRI normale - sezione sagittale media. CSF - serbatoi.
A - PIASTRA FINE SERBATOIO
B - VASCA DEL CHIASMA
C - Cisterna interpeduncolare
D - Serbatoio bypass
E - Cisterna quadrigemina
F - Cisterna cerebellopontina
G - Cisterna Pontina Prepontine pontocerebellaris Cisterna Pontina (prepontine)
H - CISTERNA CEREBELLOMIDOLARE LATERALE
I - SERBATOIO MAGNA
Immagine gentilmente concessa dal Dott. Coenraad J. Hattingh
CISTERNE DEL CERVELLO
Dal quarto ventricolo del cervello, il liquido cerebrospinale entra nelle cisterne basali attraverso i forami accoppiati di Luschka e il forame spaiato di Magendie.
Nome dei serbatoi in base alla posizione:
Nel piano sagittale:
- Cisterna soprasellare
- Cisterna prepontina nella quale passa l'arteria principale.
- Cisterna delle quattro colline
- Cisterna maggiore o basale del cervello
Nel piano assiale:
- Cisterna interpeduncolare
- La cisterna bypass collega le cisterne interpeduncolari e quadrigeminali. Anche il serbatoio di bypass ha le ali: destra e sinistra.


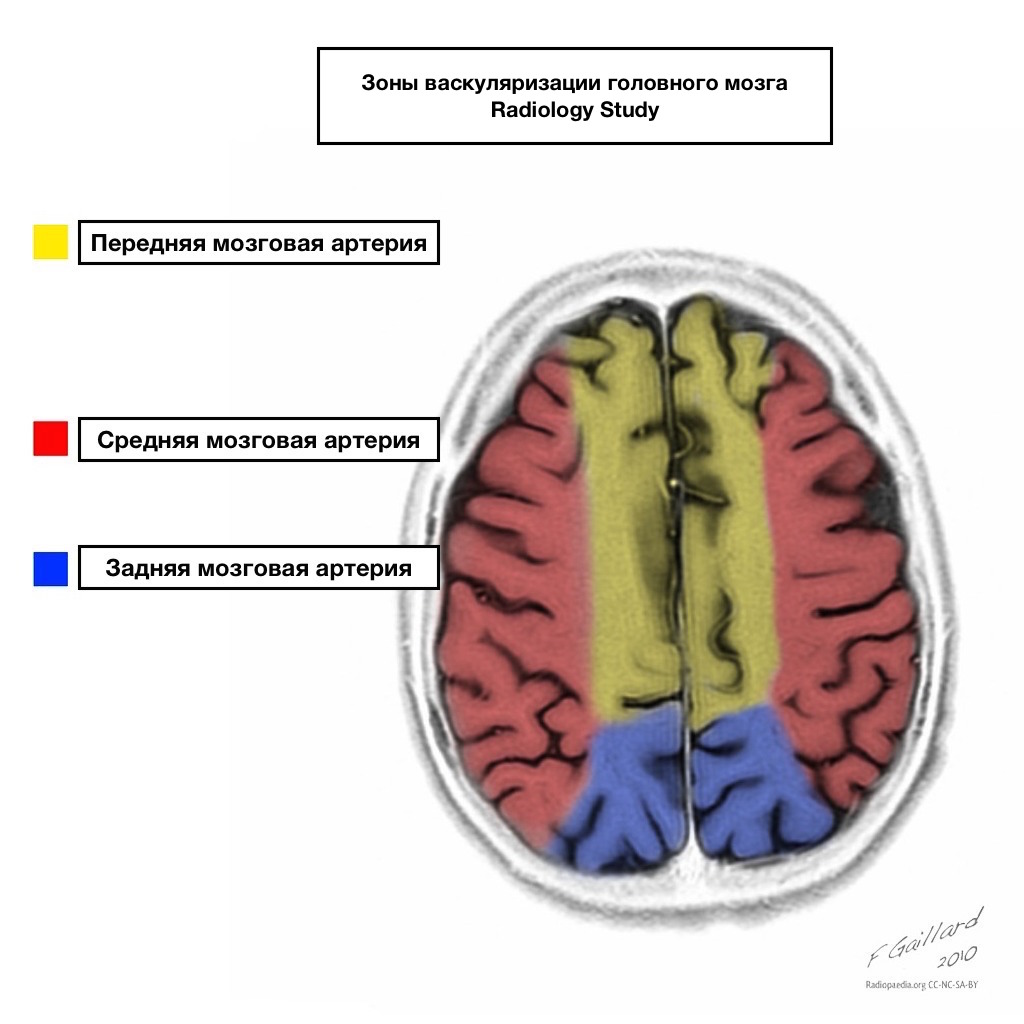







I bacini di afflusso di sangue hanno confini chiari.

Aree di afflusso sanguigno adiacente
Zone di afflusso di sangue adiacenti all'intersezione delle zone di afflusso di sangue:
Arteria cerebrale anteriore
Arteria cerebrale media
Arteria cerebrale posteriore.
Molto spesso, gli attacchi cardiaci in queste aree sono di natura emodinamica, cioè si verificano quando la pressione sanguigna diminuisce.

Meningi del cervello
Il cervello è coperto da tre membrane.
- Il guscio molle è strettamente adiacente al cervello, si estende in tutte le fessure e le scanalature e contiene vasi sanguigni. In certi punti penetra nei ventricoli del cervello e forma i plessi coroidei.
- La membrana aracnoidea o aracnoidea si trova sopra le scanalature e si diffonde da un giro all'altro.
- Il guscio duro dall'interno riveste le cavità del cranio, aderisce saldamente ad esse e forma seni venosi e processi che separano le singole strutture del cervello l'una dall'altra.
Normalmente le meningi cerebrali non vengono visualizzate alla risonanza magnetica, ma dopo la somministrazione del mezzo di contrasto viene contrastata la dura madre.

Cambiamenti nelle meningi molli.
Nella carcinomatosi leptomeningea, le immagini senza contrasto T1 e T2 mostrano un aumento del segnale proveniente dalle meningi e, dopo la somministrazione del contrasto, la visualizzazione migliora.

Meningite
I cambiamenti nelle meningi molli si riscontrano spesso anche durante i cambiamenti infiammatori, ad esempio nella leptomeningite tubercolare.

Cambiamenti nella dura madre
I cambiamenti nella dura madre si verificano con ipotensione intracranica. Con questa patologia, viene visualizzata una dura madre ispessita, che accumula intensamente contrasto. Ulteriori criteri per la diagnosi sono l'aumento delle dimensioni della ghiandola pituitaria e il prolasso delle tonsille cerebellari nel forame magno.

Cambiamenti nella dura madre si verificano anche nella carcinomatosi pachimeningea, che si manifesta con un ispessimento della dura madre con intenso accumulo di mezzo di contrasto ed edema vasogenico delle parti adiacenti del lobo frontale.
![]()
Spazi della conchiglia.
Gli spazi meningei sono gli spazi tra le membrane del cervello.
- Lo spazio subaracnoideo è lo spazio compreso tra la pia madre e la membrana aracnoidea. Normalmente, dovrebbe avere l'intensità del liquido cerebrospinale.
- Lo spazio subdurale è lo spazio compreso tra l'aracnoide e la dura madre.
- Lo spazio epidurale è lo spazio tra la dura e le ossa del cranio, che normalmente non viene visualizzato poiché la dura è fusa con le ossa del cranio.

Cambiamenti nello spazio subaracnoideo
Cambiamenti nello spazio subaracnoideo
Restringimento. Questi cambiamenti si verificano con effetti volumetrici (tumore, infarto).
Estensione. Questi cambiamenti si verificano nel periodo post-traumatico, dopo un infarto o durante l'atrofia.



Emorragie meningee
Nelle emorragie meningee siamo bravissimi a identificare le membrane.
Tipi di emorragie meningee:
Emorragia epidurale. Di solito vengono visualizzate come una lente e non si estendono oltre le suture, ma possono attraversare i seni cerebrali, che è una caratteristica distintiva dalle emorragie subdurali, che non attraversano mai i seni cerebrali.
Emorragia subdurale. La causa più comune è la rottura delle vene superficiali a seguito dello spostamento del cervello a causa di un trauma. Se in questo caso si rompe anche la membrana subaracnoidea, in questo caso il liquido cerebrospinale entra nello spazio subdurale.
Emorragia subaracnoidea. Nella modalità FLAIR viene rilevato un aumento del segnale proveniente dal liquido cerebrospinale. La causa più comune di emorragia subaracnoidea è la rottura di un aneurisma, poiché le arterie che forniscono sangue al cervello sono localizzate nello spazio subaracnoideo.

Nei processi patologici delle membrane non viene utilizzato il termine lobo, bensì regione. Ad esempio, questo paziente ha un meningioma della regione frontale.
