Systémová amyloidóza: diagnostika, diferenciálna diagnostika, liečba. Klinický priebeh a jeho hlavné prejavy
Amyloidóza- ochorenie charakterizované poruchou látkovej premeny, v dôsledku ktorej vzniká pre telo nová látka (amyloid), ktorá sa ukladá v orgánoch a narúša ich funkcie.
Amyloid je komplexný glykoproteín, v ktorom sú fibrilárne a globulárne proteíny úzko spojené s polysacharidmi. Amyloidná fibrila pozostáva z polypeptidových proteínov; amyloid obsahuje okrem fibrilárneho proteínu aj ďalší proteín – takzvanú P-zložku, ktorá je rovnaká pre všetky formy amyloidu. Zložka P je považovaná za normálny sérový proteín spojený s amyloidnými vláknami.
Amyloidóza sa môže vyskytnúť ako komplikácia akejkoľvek choroby alebo sa môže vyvinúť ako nezávislý proces.
V súčasnosti v závislosti od etiológie existuje niekoľko foriem amyloidózy, ktoré majú svoje biochemické zloženie amyloidných fibríl.
Primárna (idiopatická) amyloidóza sa vyvíja bez zjavného dôvodu a postihuje rôzne orgány (srdce, obličky, črevá, pečeň, nervový systém). Biochemickou formou primárnej amyloidózy je AL-forma, prekurzorom takéhoto amyloidu je Ig a ľahké reťazce imunoglobulínu. Podľa štruktúry amyloidu a charakteru poškodenia vnútorných orgánov je amyloidóza pri mnohopočetnom myelóme blízka primárnej (idiopatické) dávke amyloidu, ktorá je v súčasnosti rozdelená do samostatnej skupiny.
Dedičná (genetická) amyloidóza sa prejavuje prevažujúcim postihnutím obličiek, kombináciou poškodenia obličiek a nervového systému. Hereditárna amyloidóza je u nás zvyčajne spojená s periodickým ochorením, ktoré sa prenáša autozomálne dominantným spôsobom. Pri tomto ochorení môže byť jediným prejavom amyloidóza. Biochemická forma dedičnej amyloidózy je AF (prekurzor amyloidu je prealbumín). V prípade periodického ochorenia je biochemickou formou AA (prekurzorom je proteín SAA).
Získaná (sekundárna) amyloidóza sa vyskytuje najčastejšie a rozvíja sa pri reumatoidnej artritíde, Bechterevovej chorobe, tuberkulóze, chronickom hnisaní - osteomyelitíde, bronchiektázii, chronickom pľúcnom abscese, menej často pri ulceróznej kolitíde, psoriáze, lymfogranulomatóze, syfilise a iných nádoroch obličiek, Biochemická forma sekundárnej amyloidózy je AA (jej sérovým prekurzorom je proteín SAA syntetizovaný hepatocytmi).
Senilná amyloidóza je výsledkom involutívnych porúch metabolizmu bielkovín, ktoré sa nachádzajú v mozgu, pankrease a srdci. Biochemický vzorec je AS (prekurzorom je prealbumín).
Lokálna amyloidóza sa vyvíja bez zjavného dôvodu, jej biochemický vzorec je AE (prekurzor nie je známy).
Patogenéza. Známe sú len samostatné väzby patogenézy (schéma 22). Zo schémy vyplýva, že pod vplyvom génovej mutácie, ako aj vplyvom vonkajších faktorov sa imunita mení - klesá
počet T-lymfocytov. To vedie k zníženiu ich kontrolného účinku na B-systém lymfocytov. V dôsledku toho klesá počet B buniek nesúcich normálne imunoglobulíny a zvyšuje sa počet B buniek syntetizujúcich prekurzory amyloidných fibríl. Amyloidoblasty vo zvýšenom množstve produkujú fibrilárny proteín, ktorý spôsobuje syntézu amyloidu vo veľkých množstvách.
V dôsledku genetického defektu amyloidoklastov, ktorý prispieva k zníženiu ich enzymatickej aktivity, však nedochádza k dostatočnej resorpcii amyloidu. V dôsledku toho dochádza k zvýšenému ukladaniu amyloidu v tkanivách a orgánoch [Mukhin N.A., 1981].
Pri mnohopočetnom myelóme sa amyloidóza vyvíja v dôsledku zvýšenej produkcie paraproteínu plazmatickými bunkami, ktorý sa používa na tvorbu amyloidu. Zloženie amyloidu v rôznych formách amyloidózy je rôzne, čo je určené zložením proteínu amyloidných fibríl.
Pri poškodení myokardu, periférnych nervov (pozorujeme hlavne pri idiopatickej forme amyloidózy) sa amyloid ukladá okolo kolagénových vlákien väziva. Ukladanie amyloidu okolo retikulárnych vlákien sa pozoruje v léziách
obličky, črevá, pečeň, nadobličky, pankreas (s dedičnou a sekundárnou amyloidózou). Je však možná kombinácia perikolagénnych a periretikulárnych amyloidných depozitov, čo poskytuje kombinované lézie rôznych orgánov a systémov.
S ukladaním amyloidu v tkanivách klesá počet funkčných prvkov, kardiomyocytov, hepatocytov, nervových vlákien a obličkových glomerulov, čo následne vedie k rozvoju zlyhania orgánov.
Takže v srdci sa amyloid ukladá pod endokardom, v stróme a cievach myokardu a tiež pozdĺž žíl v epikarde. Súčasne sa srdce prudko zväčšuje a počet kardiomyocytov rýchlo klesá. To všetko vedie k zníženiu kontraktilnej funkcie myokardu a srdcového zlyhania, ako aj k poruchám vedenia a srdcového rytmu. V mozgu so senilnou amyloidózou sa amyloid nachádza v takzvaných senilných plakoch kôry, ciev a membrán. V koži sa amyloid ukladá v papilách a cievnych stenách, čo vedie k prudkej atrofii epidermis. V pečeni sa amyloid ukladá medzi hviezdicovými retikuloendoteliocytmi sínusových ciev, v stenách ciev, kanálikov a v spojivovom tkanive portálnych ciest. Keď sa amyloid hromadí, pečeňové bunky atrofujú.
V obličkách sa amyloid ukladá v membráne glomerulárnych kapilár a tubulov nefrónu, v mezangiu, kapilárnych slučkách a pozdĺž arteriol. Keď sa amyloid hromadí, väčšina nefrónov atrofuje, odumiera alebo je nahradená spojivovým tkanivom – objaví sa oblička scvrknutá amyloidom. Tento proces možno znázorniť ako nasledujúci diagram:
proteinúria -> nefrotický syndróm -> zlyhanie obličiek.
Podľa toho sa v klinickom obraze rozlišujú tri štádiá: 1) počiatočné (proteinurické); 2) nasadené (nefrotické); 3) terminálny (azotemický).
klinický obraz. Prejavy amyloidózy sú rôznorodé a sú určené: 1) lokalizáciou amyloidu v určitom orgáne; 2) stupeň závažnosti amyloidných usadenín v orgáne; 3) hlavné ochorenie, proti ktorému sa vyvinul amyloid (so sekundárnou formou amyloidózy).
Ťažkosti môžu nastať pri diagnostike, pretože klinické prejavy ochorenia sa prejavia až pri určitom množstve uloženého amyloidu. V tomto ohľade je „latentné“ obdobie nevyhnutné od okamihu ukladania amyloidu do objavenia sa symptómov narušeného fungovania orgánu alebo systému.
Klinický obraz je obzvlášť jasný v prípade poškodenia obličiek, najčastejšej lokalizácie amyloidných depozitov.
V prvom štádiu diagnostického vyhľadávania v počiatočnom štádiu nie je možné získať prakticky žiadne informácie naznačujúce poškodenie obličiek amyloidózou. Sťažnosti pacientov sú spojené so základným ochorením (so sekundárnou amyloidózou).
V anamnéze sú informácie o prítomnosti konkrétneho ochorenia (pľúcna tuberkulóza, osteomyelitída, reumatoidná artritída a pod.), jeho priebehu a liečbe. Tieto informácie samy o sebe neumožňujú diagnostikovať amyloidózu obličiek, ale upozorňujú lekára na túto možnosť.
V pokročilom štádiu amyloidózy sa pacienti sťažujú v dôsledku rozvoja nefrotického syndrómu na zníženie množstva moču, edém rôznej prevalencie a závažnosti, ako aj sťažnosti na slabosť, nedostatok chuti do jedla a zníženú výkonnosť. Spolu s nimi sa pri sekundárnej amyloidóze vyskytujú sťažnosti na prejav základnej choroby.
V terminálnom štádiu sú ťažkosti spôsobené rozvojom chronického zlyhania obličiek: strata chuti do jedla, nevoľnosť, vracanie (dyspeptické poruchy), bolesti hlavy, poruchy spánku (poruchy nervového systému), svrbenie kože.
V štádiu II diagnostického vyhľadávania možno v počiatočnom štádiu zistiť iba symptómy charakteristické pre základné ochorenie (so sekundárnou amyloidózou).
AT pokročilé štádium odhaliť: 1) hypostázy rôznej lokalizácie a expresivity; s výraznou retenciou tekutín v tele sa môže objaviť hydrotorax, hydroperikard, prechodný ascites; 2) arteriálna hypertenzia (vyskytuje sa u 12-20 % pacientov s amyloidózou), dilatácia a hypertrofia ľavej komory; 3) zvýšenie pečene a sleziny v dôsledku ukladania amyloidu v tkanivách (pečeň a slezina sú husté, bezbolestné, so špicatým okrajom); 4) príznaky základného ochorenia (so sekundárnou amyloidózou).
AT terminálne štádium symptómy sú určené závažnosťou zlyhania obličiek: 1) dystrofický syndróm (zmeny na koži a slizniciach); 2) serózno-kĺbový syndróm (osteoartropatia, sekundárna dna, suchá perikarditída, pleurisy); 3) arteriálna hypertenzia.
V štádiu III diagnostického hľadania amyloidózy sa získajú najvýznamnejšie informácie na stanovenie diagnózy, ktoré možno rozdeliť do nasledujúcich skupín: 1) močový syndróm; 2) porušenie metabolizmu bielkovín a lipidov; 3) detekcia usadenín amyloidných hmôt.
močový syndróm: 1) proteinúria - najdôležitejší príznak amyloidózy, vyvíja sa vo všetkých formách, najčastejšie však pri sekundárnej amyloidóze. Proteinúria býva výrazná, denne sa uvoľní 2-20 g bielkovín, ktorých hlavnú časť tvorí albumín. V menšom množstve sa vylučujú globulíny, močom je možné vylúčiť sérový amyloidový prekurzor (bielkovina SAA). V terminálnom štádiu proteinúria pretrváva. V moči sa dajú zistiť a- a najmä y-glykoproteíny.
Podľa stupňa proteinúrie sa nachádzajú hyalínové a menej často granulované odliatky. Zriedkavo je diagnostikovaná mikrohematúria alebo leukocytúria, ale jej závažnosť nezodpovedá stupňu proteinúrie (ako sa pozoruje pri glomerulonefritíde). Stupeň porúch metabolizmu lipidov pri amyloidóze zodpovedá lipoidúrii s prítomnosťou dvojlomných kryštálov v močovom sedimente.
Poruchy metabolizmu bielkovín a lipidov: 1) hypoproteinémia v kombinácii s hypoalbuminémiou a hyper-<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
Dôležité pre diagnózu je detekcia amyloidných hmôt v orgánoch a tkanivách: v pečeni (50 % prípadov), slezine (s punkčnou biopsiou), sliznici ďasien a konečníka.
V počiatočnom štádiu (proteinurická) biopsia sliznice ďasien často dáva negatívny výsledok a konečník - pozitívny výsledok; v pokročilom štádiu (nefrotické) v prvom prípade sú výsledky pozitívne u polovice pacientov av druhom - ešte častejšie.
Napokon, pri chronickom zlyhaní obličiek sú bioptické údaje tkaniva ďasien vo viac ako polovici prípadov pozitívne a údaje z biopsie rektálnej sliznice takmer vo všetkých prípadoch. Pre pokročilý proces by sa preto mala odporučiť biopsia sliznice ďasien a biopsia konečníka - v akomkoľvek štádiu amyloidózy.
Pri podozrení na idiopatickú amyloidózu (vyskytuje sa častejšie pri poškodení srdca, periférnych nervov, menej často - obličiek) je vhodné najskôr vykonať biopsiu sliznice ďasien a v prípade sekundárnej (získanej) amyloidózy a jej dedičné formy (vyskytujú sa s prevažujúcim poškodením obličiek) - biopsia sliznice konečníka.
Množstvo ďalších štúdií pomáha: 1) objasniť diagnózu ochorenia, proti ktorému sa vyvinula amyloidóza; 2) posúdiť funkčný stav obličiek (Rebergov test, Zimnitského test, hladina kreatinínu v krvi).
Prietok. Klinický obraz renálnej amyloidózy má znaky, ktoré ju odlišujú od poškodenia obličiek rôzneho pôvodu: 1) nefrotický syndróm vzniká postupne a často po dlhom štádiu proteinúrie, je charakterizovaný pretrvávajúcim priebehom, edémy sú často rezistentné na rôzne diuretiká. Pri CGN sa nefrotický syndróm vyskytuje spravidla už na začiatku ochorenia a často sa opakuje v budúcnosti; 2) arteriálna hypertenzia sa pozoruje zriedkavo, dokonca aj v štádiu chronického zlyhania obličiek; 3) pri primárnej amyloidóze prebieha chronické zlyhanie obličiek benígnejšie, na rozdiel od sekundárnej amyloidózy alebo CGN (kvôli menšej závažnosti glomerulárneho poškodenia v porovnaní so sekundárnymi formami amyloidózy); 4) priebeh sekundárnej amyloidózy do značnej miery závisí od základného ochorenia, s častými exacerbáciami, pri ktorých je možná významná progresia amyloidózy.
Komplikácie. S amyloidózou sa v 2-5% prípadov vyvinie:
1) trombóza obličkových žíl (so sekundárnou amyloidózou), ktorá sa prejavuje
hematúria a bolesť v bedrovej oblasti, zvýšenie bielkovín
ria a pokles diurézy;
2) interkurentná infekcia;
3) fibrinózno-hnisavá peritonitída, ktorej výskyt je sprevádzaný
dochádza k prudkému nárastu ascitu.
Diagnostika. Klinické prejavy amyloidózy sú nešpecifické. Každý z príznakov (edém, proteinúria, arteriálna hypertenzia) sa môže vyskytnúť pri rôznych ochoreniach obličiek. Jedinou metódou spoľahlivej diagnózy amyloidózy je biopsia orgánu (obličky, pečene, rektálnej sliznice alebo ďasien), ale nie vždy je to možné. Preto je vo väčšine prípadov potrebné zamerať sa na klinické prejavy patologického procesu.
Prítomnosť ochorenia, pri ktorom sekundárne
amyloidóza (klinické alebo anamnestické príznaky).
Nástup a progresia proteinúrie alebo nástup
nefrotický syndróm.
Neexistuje žiadna choroba, pri ktorej by sa mohla vyvinúť amyloidóza,
existuje však proteinúria alebo nefrotický syndróm.
Prítomnosť pretrvávajúceho závažného srdcového zlyhania, syndróm nie je
dostatočná absorpcia, polyneuropatia (ak zároveň posledná
Je ťažké vysvetliť tieto tri syndrómy inými dôvodmi).
Prítomnosť amyloidózy možno predpokladať pri nasledujúcich laboratórnych príznakoch nefrotického syndrómu (ktorý, ako viete, sa môže vyvinúť pri iných ochoreniach obličiek):
a) ťažká dysproteinémia + hypoalbuminémia + hyper-sg- a gi-
pergamaglobulinémia;
b) zvýšenie hladiny ag-glykoproteínu, p-lipoproteínov;
c) výskyt a- a najmä y-glykoproteínov a a-lipopro- v moči
teidy.
Vo všetkých prípadoch sa pravdepodobnosť vzniku amyloidózy zvyšuje s detekciou hepato- a splenomegálie, ako aj zmien v srdci charakteristických pre amyloidózu (v takýchto prípadoch hovoríme o idiopatickej generalizovanej amyloidóze).
Diagnózu renálnej amyloidózy je preto možné s dostatočnou istotou stanoviť v pokročilom (nefrotickom) alebo terminálnom štádiu, kým v počiatočnom (proteinurickom) štádiu je to oveľa ťažšie. V týchto prípadoch treba odlíšiť prechodnú alebo trvalú proteinúriu od glomerulonefritídy (akútnej, chronickej). Toto by malo brať do úvahy:
1) pomalšia progresia poškodenia obličiek pri amyloide
dávka;
2) absencia jasného spojenia s prechladnutím pri amyloidóze
niami;
3) stála prítomnosť mikrohematúrie pri glomerulonefritíde (s
amyloidóza v 20 % prípadov).
Niekedy môže byť správna diagnóza stanovená až po období dlhého pozorovania pacienta. Problém je vyriešený oveľa rýchlejšie, ak je možné vykonať punkčnú biopsiu obličky.
Formulácia podrobnej klinickej diagnózy amyloidóza berie do úvahy nasledujúce zložky: 1) forma amyloidózy; 2) štádium amyloidózy (proteinurická, nefrotická, terminálna); 3) funkčný stav obličiek (neprítomnosť alebo prítomnosť zlyhania obličiek, stupeň jeho závažnosti); 4) základné ochorenie (so sekundárnou amyloidózou); 5) stav iných orgánov (srdce, pečeň, nervový systém atď.) pri idiopatickej (primárnej) amyloidóze.
Liečba. Problém liečby amyloidózy zostáva stále nevyriešený, pretože príčiny vedúce k zvýšenej amyloidogenéze a nedostatočnej jej resorpcii neboli objasnené. Napriek tomu je možné vykonať sériu terapeutických opatrení, ktoré zlepšujú stav pacienta. V súčasnosti sa liečba pacienta s amyloidózou vykonáva s prihliadnutím na: 1) vplyv na základné ochorenie, proti ktorému sa amyloidóza vyvinula
(sekundárne); 2) vplyv na mechanizmy patogenézy; 3) účinky na hlavné klinické syndrómy.
Vplyv na základnú chorobu, proti ktorej sa vyvíja
sekundárna amyloidóza, je potrebné vzhľadom na to, že často
strie alebo vysoká aktivita patologického procesu vedú k
progresia amyloidózy.
Tento vplyv je nasledujúci:
a) pri chronických infekciách (tuberkulóza, syfilis)
dlhodobá špecifická terapia;
b) pri chronických nešpecifických ochoreniach pľúc - kom
plexová terapia antibiotikami, drenáž priedušiek a
ak je to potrebné, a chirurgická intervencia (napríklad s chronickým
absces pľúc);
c) so systémovými ochoreniami spojivového tkaniva, napríklad s
reumatoidná artritída, je indikovaná komplexná liečba, vrátane
hodnota základných prípravkov (D-penicilamín, soli zlata, amino-
nolínové lieky).
Vplyv na mechanizmy patogenézy zahŕňa pokles
syntéza amyloidu:
a) denný príjem 80-120 g surovej pečene počas 6-12 mesiacov
vedie k zníženiu proteinúrie, zníženiu veľkosti pečene
ani slezina;
b) aminochinolínové prípravky (hingamin alebo delagil, podľa
0,25 - 0,5 g denne po mnoho mesiacov a dokonca rokov)
no postup procesu. Zdá sa, že tieto prostriedky vplyvu
yut o syntéze amyloidných fibríl. Liečba je len účinná
v počiatočných štádiách amyloidózy; v pokročilom procese
(detailný nefrotický syndróm, renálna insuficiencia)
ness) vymenovanie týchto liekov je nepraktické;
c) s rozvojom amyloidózy v dôsledku periodického ochorenia re
odporúča sa kolchicín;
d) pri primárnej amyloidóze sa predpisuje aj melfalan, ktorý inhibuje
funkcie niektorých klonov lymfocytov, najmä syn
typizácia imunoglobulínov s ľahkým reťazcom zapojených do
tvorba amyloidnej fibrily (súvisí to aj s
amyloidóza, ktorá sa vyvíja pri mnohopočetnom myelóme).
Vplyv na hlavné klinické syndrómy zahŕňa
odstránenie edému, hypertenzie, ako aj opatrenia zamerané na
boj proti rozvoju zlyhania obličiek:
a) s rozvojom nefrotického syndrómu a ťažkým edémom
potrebný je dostatočný obsah bielkovín v potrave, zníženie v
prevarená soľ, ako aj zavedenie celej krvi alebo erytrocytov-
noy hmotnosť (najmä v prítomnosti anémie), starostlivá aplikácia
užívanie diuretík;
b) arteriálna hypertenzia sa zriedkavo vyskytuje pri amyloidóze,
keď však dosiahne vysoké čísla, je potrebné vymenovať
užívanie antihypertenzív rôznych typov;
c) s rozvojom zlyhania obličiek sa liečba uskutočňuje podľa všeobecne uznávaného plánu (obmedzenie bielkovín v potrave, dostatočný príjem tekutín, úprava metabolizmu minerálov). Pri zlyhaní obličiek v dôsledku amyloidózy je možné použiť hemodialýzu a transplantáciu obličky.
Predpoveď. Je ťažké stanoviť trvanie proteinurického obdobia, avšak po jeho zistení sa zvyčajne po 3 rokoch vyvinie edém, proti ktorému sa rýchlo rozvinie CRF. To všetko robí predpoveď dosť vážnou.
Prevencia. Pri idiopatickej a genetickej amyloidóze nie sú opatrenia primárnej prevencie známe. Pri sekundárnej amyloidóze spočíva prevencia v liečbe ochorení vedúcich k rozvoju amyloidózy.
RCHD (Republikové centrum pre rozvoj zdravia Ministerstva zdravotníctva Kazašskej republiky)
Verzia: Klinické protokoly Ministerstva zdravotníctva Kazašskej republiky - 2016
Amyloidóza (E85)
Nefrológia
všeobecné informácie
Stručný opis
Schválené
Spoločná komisia pre kvalitu zdravotníckych služieb
Ministerstvo zdravotníctva a sociálneho rozvoja Kazašskej republiky
zo dňa 13.10.2016
Protokol č. 13
Amyloidóza- skupina ochorení, ktorých charakteristickým znakom je ukladanie v tkanivách a orgánoch fibrilárneho glykoproteínu - amyloidu.
Korelácia medzi kódmi ICD-10 a ICD-9
| ICD-10 | ICD-9 | ||
| Kód | názov | Kód | názov |
| E85 | Amyloidóza |
55.23 99.76 |
Uzavretá [perkutánna] [punkčná] biopsia obličky. Hemodialýza. Terapeutická plazmaferéza Extrakorporálna imunoadsorpcia |
| E85.0 | Dedičná familiárna amyloidóza bez neuropatie | ||
| E85.1 | Neuropatická dedičná amyloidóza | ||
| E85.2 | Nešpecifikovaná dedičná amyloidóza | ||
| E85.3 | Sekundárna systémová amyloidóza | ||
| E85.4 | Obmedzená amyloidóza | ||
| E85.8 | Iné formy amyloidózy | ||
| E85.9 | Amyloidóza, bližšie neurčená | ||
Dátum vypracovania/revízie protokolu: 2016.
Používatelia protokolu: všeobecných lekárov, terapeutov, hematológov, nefrológov.
Stupnica úrovne dôkazov:
| ALE | Vysokokvalitná metaanalýza, systematický prehľad RCT alebo veľké RCT s veľmi nízkou pravdepodobnosťou (++) zaujatosti, ktorých výsledky možno zovšeobecniť na vhodnú populáciu. |
| AT | Vysokokvalitné (++) systematické preskúmanie kohortových alebo prípadovo-kontrolných štúdií alebo Vysokokvalitné (++) kohortové alebo prípadové kontrolné štúdie s veľmi nízkym rizikom zaujatosti alebo RCT s nízkym (+) rizikom zaujatosti, výsledky ktoré možno zovšeobecniť na príslušnú populáciu . |
| OD |
Kohorta alebo prípad-kontrola alebo kontrolovaná štúdia bez randomizácie s nízkym rizikom zaujatosti (+). Výsledky ktorých možno zovšeobecniť na príslušnú populáciu alebo RCT s veľmi nízkym alebo nízkym rizikom zaujatosti (++ alebo +), ktorých výsledky nemožno priamo zovšeobecniť na príslušnú populáciu. |
| D | Opis série prípadov alebo nekontrolovanej štúdie alebo znaleckého posudku. |
Klasifikácia
Typy amyloidov a zodpovedajúce formy amyloidózy:
|
Proteín amyloid |
Prekurzorový proteín | Klinická forma amyloidózy |
| AA | SAA proteín | Sekundárna amyloidóza pri chronických zápalových ochoreniach, vrátane periodického ochorenia a Muckle-Wellsovho syndrómu |
| AL | λ, κ-ľahké reťazce imunoglobulínov | Amyloidóza pri dyskráziách plazmatických buniek - idiopatická, pri mnohopočetnom myelóme a Waldenströmovej makroglobulinémii |
| ATTR | transtyretín | Rodinné formy polyneuropatickej, kardiopatickej a inej amyloidózy, systémová senilná amyloidóza |
| Ap2M | β2-mikroglobulín | dialyzačná amyloidóza |
| AGel | Gelsolin | Fínska familiárna amyloidná polyneuropatia |
| AApoAI | Apolipoproteín A-I | Amyloidná polyneuropatia (typ III, podľa van Allena, 1956) |
| AFib | fibrinogén | amyloidná nefropatia |
| Ap | β-proteín | Alzheimerova choroba, Downov syndróm, dedičné cerebrálne krvácania s amyloidózou, Holandsko |
| APrP Scr | Priónový proteín | Creutzfeldt-Jakobova choroba, Gerstmann-Straussler-Scheinkerova choroba |
| AANF | Predsieňový natriuretický faktor | Izolovaná atriálna amyloidóza |
| AIAPP | Amylin | Izolovaná amyloidóza v Langerhansových ostrovčekoch pri diabetes mellitus typu II, inzulinóm |
| ACal | Prokalcitonín | Pre medulárnu rakovinu štítnej žľazy |
| ACys | Cystatín C | Dedičné cerebrálne krvácania s amyloidózou, Island |
Klinická klasifikácia amyloidózy
primárna amyloidóza:
vyskytujúce sa bez zjavného dôvodu;
Súvisí s mnohopočetným myelómom
sekundárna amyloidóza:
pri chronických infekciách;
pri reumatoidnej artritíde a iných ochoreniach spojivového tkaniva;
pri onkologických ochoreniach;
familiárna (dedičná) amyloidóza:
v prípade periodickej choroby;
portugalský variant a iné formy familiárnej amyloidózy;
senilná amyloidóza
lokálna amyloidóza
dedičná amyloidóza:
neuropatické
s poškodením dolných končatín: portugalské, japonské, švédske a iné typy;
s léziami horných končatín: typy Švajčiarsko-Indiana, Nemecko-Maryland;
nefropatické:
Pravidelné ochorenie
horúčka a bolesť brucha u Švédov a Sicílčanov;
kombinácia vyrážky, hluchoty a poškodenia obličiek;
Poškodenie obličiek v kombinácii s arteriálnou hypertenziou;
kardiomyopatické:
dánsky - progresívne srdcové zlyhanie;
· mexicko-americký - syndróm chorého sínusu, zástava predsiení;
zmiešané:
fínsky - dystrofia rohovky a poškodenie hlavových nervov;
mozgové príhody.
Klinické štádiá amyloidózy obličiek
| Etapa | Klinický prejav |
| 1 | Predklinické alebo latentné (asymptomatické) štádium - v intermediárnej zóne je prítomný amyloid a pozdĺž priamych ciev pyramíd sa vyvíjajú edémy a ložiská sklerózy. Štádium trvá 3-5 alebo viac rokov. V tomto období dominujú reaktívnej amyloidóze klinické prejavy základného ochorenia (napr. hnisavý proces v pľúcach, tuberkulóza, reumatoidná artritída atď.). |
| 2 | Proteinurické (albuminurické) štádium – amyloid sa objavuje predovšetkým v mezangiu, v slučkách kapilár, v pyramídach a kortikálnej substancii glomerulov, v cievach. Rozvíja sa skleróza a atrofia nefrónov, hyperémia a lymfostáza. Obličky sú zväčšené a husté, matnej šedo-ružovej farby. Proteinúria na začiatku je mierne vyjadrená, môže byť dokonca prechodná po určitú dobu, klesá a stúpa, ale potom sa stáva perzistentnou (štádium intermitentnej proteinúrie). Niektorí vedci rozlišujú v tomto štádiu dve obdobia: selektívnu a neselektívnu proteinúriu. Trvanie etapy je od 10 do 13 rokov. |
| 3 | Nefrotické (edematózne, edematózno-hypotonické) štádium - amyloid-lipoidná nefróza - amyloid vo všetkých častiach nefrónu. Existuje skleróza a amyloidóza drene, ale kortikálna vrstva bez výrazných sklerotických zmien. Trvanie etapy je až 6 rokov. V proteinurickom aj nefrotickom štádiu sú obličky zväčšené, husté (veľká mazová oblička). Klinicky sa toto štádium prejavuje klasickým nefrotickým syndrómom so všetkými jeho znakmi: s rozvojom masívnej proteinúrie (so stratou bielkovín v moči viac ako 3-5 gramov denne), hypoproteinémiou s hypoalbuminémiou, hypercholesterolémiou, lipidúriou s. edém až do stupňa anasarky. Hyalínové odliatky sa nachádzajú v močovom sedimente a so zvyšujúcou sa proteinúriou sa nachádzajú granulované odliatky. Možná mikro- a makrohematúria, leukocytúria bez príznakov pyelonefritídy. |
| 4 | Uremická (terminálna, azotemichesky) fáza - amyloidná vráskavá oblička - zmenšená, hustá, zjazvená oblička. Chronické zlyhanie obličiek sa len málo líši od zlyhania iných ochorení obličiek. Predpokladá sa, že na rozdiel od glomerulonefritídy, pri ktorej nástup CRF vyskytujúci sa s polyúriou môže viesť k aspoň čiastočnej konvergencii edému, pri amyloidóze sa azotémia vyvíja na pozadí nízkeho krvného tlaku a nefrotického syndrómu. |
Diagnostika (ambulancia)
DIAGNOSTIKA NA AMBULANCOVEJ ÚROVNI
Diagnostické kritériá
Sťažnosti:
Slabosť, zvýšená únava;
· bolesť hlavy;
opuch nôh, rúk a tváre;
vysoký krvný tlak;
nevoľnosť, hnačka (hnačka);
bolesť v oblasti srdca;
bolesť svalov.
Anamnéza:
· strata váhy;
Prítomnosť monoklonálnej gamapatie neznámeho pôvodu;
chronické zápalové (hnisavé) ochorenia;
chronické infekcie;
dedičnosť.
Fyzikálne vyšetrenie
Všeobecná kontrola:
Periorbitálna purpura (pozorovaná v 15% prípadov);
makroglosia je charakteristická pre primárnu amyloidózu (AL);
Dýchavičnosť počas fyzickej námahy (pozorovaná asi u 40% pacientov);
znak ramena (periartikulárna infiltrácia amyloidu vedie k falošnej hypertrofii a zväčšeniu objemu svalov ramenného pletenca a stehna).
Auskultácia:
možná prítomnosť srdcových arytmií.
Palpácia:
opuchy dolných končatín v dôsledku hypoalbuminémie a nefrotického syndrómu, ako aj v dôsledku stagnácie systémového obehu v dôsledku reštriktívnej kardiomyopatie (pozorované v 50% prípadov);
zväčšenie pečene a sleziny;
parestézia (pozorovaná asi u 15 % pacientov);
Spastická bolesť v gastrointestinálnom trakte;
Môže dôjsť k zvýšeniu submandibulárnych slinných žliaz.
Laboratórny výskum:
Kompletný krvný obraz - anémia, leukocytóza, zvýšená ESR;
všeobecný rozbor moču - proteinúria, mikrohematúria, aseptická leukocytúria;
Biochemický krvný test (celkový proteín, albumín, Na, Ca, cholesterol, cukor v krvnom sére) - hypoproteinémia (v dôsledku hypoalbuminémie), hyperglobulinémia, hyponatrémia, hypoprotrombinémia, hypokalciémia, hypercholesterolémia.
Ultrazvuk brušných orgánov a obličiek - vizualizujú sa zväčšené zhutnené obličky (veľké mastné obličky).

Diagnostika (nemocnica)
DIAGNOSTIKA NA STACIONÁRNEJ ÚROVNI
Diagnostické kritériá na úrovni nemocnice
Sťažnosti a anamnéza: pozri ambulantnú úroveň.
Fyzikálne vyšetrenie: pozri ambulantnú úroveň.
Laboratórny výskum:
| diagnostický test | Výsledok |
|
Imunofixácia séra Test je pozitívny u 60 % pacientov s amyloidózou s ľahkým reťazcom imunoglobulínu (AL) (6). |
|
|
Imunofixácia moču Test je pozitívny u 80 % pacientov s AL amyloidózou (6). Detekcia proteínu ľahkého reťazca v moči naznačuje prítomnosť mnohopočetného myelómu a amyloidózy. |
Prítomnosť monoklonálneho proteínu |
|
Testovanie imunoglobulínov s voľným ľahkým reťazcom v sére Tento relatívne nový test má veľmi vysokú senzitivitu (> 95 %) na diagnostiku AL amyloidózy (10). Bežne sa používajú komerčne dostupné antiséra proti ľahkým reťazcom imunoglobulínov, AA a transtyretínu, ale nemusia mať dostatočnú špecifickosť a citlivosť. V mnohých prípadoch je na určenie základného typu amyloidu potrebná hmotnostná spektroskopia a imunoelektrónová mikroskopia. |
Abnormálny pomer kappa lambda |
|
Biopsia kostnej drene Biopsia kostnej drene sa vykonáva u všetkých pacientov s podozrením na amyloidózu s ľahkým reťazcom a je výborným zdrojom tkaniva na diagnostiku každého pacienta s podozrením na amyloidózu. |
Prítomnosť klonálnych plazmatických buniek |
Inštrumentálny výskum:
Ultrazvuk brušných orgánov a obličiek - vizualizujú sa zväčšené zhutnené obličky (veľké mastné obličky).
Diagnostický algoritmus pre renálnu amyloidózu
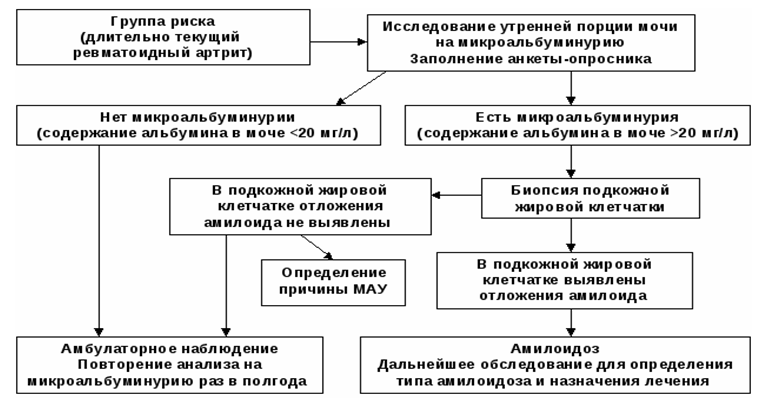
Zoznam hlavných diagnostických opatrení:
· všeobecný rozbor krvi;
· všeobecný rozbor moču;
biochemická analýza krvi (celkový proteín, albumín, Na, Ca, cholesterol, cukor v krvnom sére);
sérová imunofixácia;
imunofixácia moču;
Vyšetrovanie voľných imunoglobulínov ľahkého reťazca v sére;
biopsia kostnej drene.
Ultrazvuk brušných orgánov a obličiek.
Zoznam dodatočných diagnostických opatrení
Laboratórny výskum:
| diagnostický test | Výsledok |
|
Biopsia tkaniva: Pre diagnostiku amyloidózy je potrebné, aby sa depozity v tkanivách v bioptickom materiáli farbili pozitívne na Kongo červeň (11). Jasne zelený dvojlom možno vidieť, keď je materiál Kongo zafarbený na červeno pod polarizovaným svetlom. Bioptický materiál možno získať zo sliznice pier, kože, ďasien, podkožného tuku, kostnej drene, nervov, konečníka, obličiek, pečene alebo srdca. Ložiská sú vždy umiestnené extracelulárne a sú amorfné. |
pozitívny - zelený dvojlom pri farbení konžskou červenou |
|
Imunohistologické štúdie amyloidných depozitov: Umožňujú vám rozpoznať rôzne formy systémovej amyloidózy. |
antisérum proti imunoglobulínu s ľahkým reťazcom, AA a transtyretínu |
|
Hmotnostná spektroskopia: Poskytuje analýzu zloženia amyloidných proteínov. V súčasnosti je zlatým štandardom diagnostiky typu amyloidózy. |
potvrdzuje typ proteínu |
|
Imunoelektrónová mikroskopia: Všetky formy amyloidu pod elektrónovým mikroskopom sú vláknité, tuhé a nerozvetvené. |
amyloidy majú fibrilárny vzhľad a sú tuhé a nerozvetvené. |
|
Genetické testovanie: Genetické vyšetrenie je povinné na vylúčenie dedičnej amyloidózy v prípade pochybných výsledkov testu na detekciu proteínov monoklonálneho imunoglobulínu/ľahkého reťazca voľného amyloidu. Gény možno skúmať priamym sekvenovaním a zahŕňajú nasledujúce gény: TTR, fibrinogén, apolipoproteín A1, lyzozým, MEFV (stredomorská horúčka) a receptor 1 faktora nekrózy nádorov (TNFR1 alebo TNFRSF1A). MEFV a TNFRSF1A sú dedičné syndrómy periodickej horúčky (tj potenciálne príčiny AA amyloidózy) a nie sú to dedičné amyloidózy ako také. |
pozitívne |
|
Scintigrafický sken sérového amyloidu P (SAP): V posledných rokoch sa v klinickej praxi na posúdenie distribúcie amyloidu v organizme začala používať scintigrafia sérového P-komponentu (SAP) značeného jódom. |
absorpcie v miestach ukladania amyloidu |
|
Všeobecná analýza krvi: Anémia sa vyskytuje hlavne u pacientov s renálnou insuficienciou alebo krvácaním z gastrointestinálneho traktu. Trombocytémia je spojená s postihnutím pečene a hypersplenizmom. |
zvyčajne normálne |
|
Biochemický krvný test (pečeňové a obličkové testy), indikátory metabolického stavu): Amyloidóza pečene je charakterizovaná zvýšenými hladinami alkalickej fosfatázy. Väčšina pacientov v počiatočných štádiách renálnej amyloidózy si zachováva klírens kreatinínu, ale môžu sa vyskytnúť významné stupne hypoalbuminémie v dôsledku straty bielkovín v moči (nefrotický syndróm). |
Nízky albumín; zvýšenie alkalickej fosfatázy |
|
Denná proteinúria (zachytenie moču za 24 hodín): Vylučovanie albumínu > 1 g/deň u pacientov s amyloidózou naznačuje poškodenie obličiek (renálna amyloidóza). Pri hladine proteinúrie > 3 g/deň vzniká nefrotický syndróm. |
zvýšená bielkovina v moči |
|
Hladina troponínu v sére: Citlivý test na určenie poškodenia myokardu. Pacienti s detekovateľnou hladinou troponínu majú horšiu prognózu ako pacienti bez troponínu (12). |
zvýšené |
|
Natriuretický peptid typu B: Citlivá diagnostická štúdia na prítomnosť distenzie myokardu a CHF. Ukázalo sa, že má dôležitú prognostickú hodnotu pri stanovení srdcovej amyloidózy (13). Na úrovni natriuretického peptidu typu B > 300 ng/l (> 300 pg/ml) naznačuje postihnutie myokardu amyloidom (10). Pacienti s<170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с >170 ng/l (> 170 pg/ml). |
zvýšené |
|
beta-2 mikroglobulíny: Je to prediktor prežitia u pacientov s amyloidózou. Pri hladine beta-2-mikroglobulínu > 2,7 mg/l je prognóza nepriaznivá (14). |
zvýšené |
Inštrumentálny výskum:
|
EKG: Má sa vykonať u všetkých pacientov ako súčasť hodnotenia srdcového postihnutia. |
porucha vedenia srdca |
|
Echokardiogram (EchoCG): Klinické príznaky srdcového zlyhania u pacientov so srdcovou amyloidózou sa pozorujú od 22 % do 34 % (7). Echokardiografia odhaľuje vysoký výskyt ukladania amyloidu u pacientov s minimálnymi symptómami (asi 50 % prípadov AL má postihnutie srdca). V poslednom štádiu dochádza k poklesu ejekčnej frakcie. |
diastolická dysfunkcia, zhrubnutie medzikomorového septa, znížená ejekčná frakcia |
|
Dopplerovská ozvena s napätím: Indikátor stupňa infiltrácie amyloidu v myokarde. Má vysokú citlivosť pri zisťovaní abnormalít, keď neexistuje arteriálna hypertenzia alebo chlopňové ochorenie srdca. Natiahnutie myokardu je definované ako percentuálna zmena dĺžky vlákna myokardu na jednotku dĺžky a rýchlosť závisí od trvania natiahnutia (15–16). |
Zníženie pozdĺžnej kontrakcie a natiahnutia myokardu; obmedzenie plnenia komôr |
|
MRI srdca: Relaxometria pomocou magnetickej rezonancie zlepšuje spoľahlivosť MRI diagnostiky a pomáha odlíšiť srdcovú amyloidózu od hypertrofickej kardiomyopatie. |
významne zvýšili relaxačné časy T1 a T2 v porovnaní s vekovými kontrolami |
Odlišná diagnóza
Diferenciálna diagnostika amyloidózy obličiek
| Štát | Rozlíšiteľné znaky/príznaky | Diferenciovateľné testy |
| Hypertrofická kardiomyopatia | Je klinicky ťažké rozlíšiť HCM od srdcovej amyloidózy. |
EchoCG je diagnostickým kritériom pre HCM, kde je zistená asymetrická hypertrofia medzikomorového septa; Dopplerovská echo s napätím, používaná na vylúčenie príznakov amyloidózy, nenaznačuje typické reštriktívne zmeny výplne zistené pri amyloidóze; MRI dokáže rozlíšiť 2 syndrómy |
| Membranózna glomerulopatia | Klinicky identické prejavy u pacientov s nefrotickým syndrómom. | Renálna biopsia sa nezafarbí konžskou červenou. |
| Neuropatia spojená s monoklonálnou gamapatiou neznámeho pôvodu (MGNG). | Pacienti nemajú významný stupeň proteinúrie, hepatomegálie alebo kardiomyopatie. | biopsia surálneho nervu sa nezafarbí konžskou červenou. |
| mnohopočetný myelóm | Bolesť kostí, príznaky anémie a zlyhania obličiek. |
Obyčajné röntgenové snímky ukazujú lytické kostné lézie, kompresné zlomeniny, difúznu osteoporózu; nízky Hb; zlyhanie obličiek. |
| nefrotický syndróm | Denná proteinúria viac ako 3,5 g/deň, edém, hypoalbuminémia, dyslipidémia | pozri KP "Nefrotický syndróm" |
Lekárska turistika
Získajte liečbu v Kórei, Izraeli, Nemecku, USA
Lekárska turistika
Nechajte si poradiť o zdravotnej turistike
Liečba
Pozor!
- Samoliečbou môžete spôsobiť nenapraviteľné poškodenie zdravia.
- Informácie zverejnené na webovej stránke MedElement nemôžu a nemali by nahradiť osobnú lekársku konzultáciu. Ak máte nejaké ochorenia alebo príznaky, ktoré vás trápia, určite sa obráťte na zdravotnícke zariadenia.
- Výber liekov a ich dávkovanie treba konzultovať s odborníkom. Len lekár môže predpísať správny liek a jeho dávkovanie s prihliadnutím na chorobu a stav tela pacienta.
- Webová stránka MedElement je len informačný a referenčný zdroj. Informácie zverejnené na tejto stránke by sa nemali používať na svojvoľnú zmenu lekárskych predpisov.
- Redakcia MedElement nezodpovedá za žiadne škody na zdraví alebo materiálne škody vyplývajúce z používania tejto stránky.
- celkové, systémové ochorenie organizmu, pri ktorom dochádza k ukladaniu špecifického glykoproteínu (amyloidu) v orgánoch a tkanivách s poškodenou ich funkciou. Pri amyloidóze môžu byť postihnuté obličky (nefrotický syndróm, edematózny syndróm), srdce (zlyhávanie srdca, arytmie), gastrointestinálny trakt, pohybový aparát, koža. Možno vývoj polyserozitídy, hemoragického syndrómu, duševných porúch. Spoľahlivú diagnostiku amyloidózy uľahčuje detekcia amyloidu v bioptických vzorkách postihnutých tkanív. Na liečbu amyloidózy sa uskutočňuje imunosupresívna a symptomatická terapia; podľa indikácií - peritoneálna dialýza, transplantácia obličiek a pečene.
ICD-10
E85

Všeobecné informácie
Amyloidóza je ochorenie zo skupiny systémových dysproteinóz, ku ktorému dochádza pri tvorbe a akumulácii komplexnej proteín-polysacharidovej zlúčeniny - amyloidu v tkanivách. Výskyt amyloidózy vo svete je do značnej miery určený geograficky: napríklad periodické ochorenie je bežnejšie v krajinách Stredomoria; amyloidná polyneuropatia - v Japonsku, Taliansku, Švédsku, Portugalsku atď. Priemerná frekvencia amyloidózy v populácii je 1 prípad na 50 tisíc obyvateľov. Choroba sa zvyčajne vyvíja u ľudí starších ako 50-60 rokov. Vzhľadom na to, že pri amyloidóze sú postihnuté takmer všetky orgánové systémy, chorobu študujú rôzne medicínske odbory: reumatológia, urológia, kardiológia, gastroenterológia, neurológia atď.
Príčiny amyloidózy
Etiológia primárnej amyloidózy nie je úplne objasnená. Zároveň je známe, že sekundárna amyloidóza býva spojená s chronickými infekčnými (tuberkulóza, syfilis, aktinomykóza) a purulentno-zápalovými ochoreniami (osteomyelitída, bronchiektázia, bakteriálna endokarditída atď.), menej často s nádorovými procesmi (lymfogranulomatóza, leukémia , viscerálna rakovina).orgány). Reaktívna amyloidóza sa môže vyvinúť u pacientov s aterosklerózou, psoriázou, reumatológiou (reumatoidná artritída, ankylozujúca spondylitída), chronickým zápalom (ulcerózna kolitída, Crohnova choroba), multisystémovými léziami (Whippleova choroba, sarkoidóza). Z faktorov, ktoré sa podieľajú na vzniku amyloidózy, majú prvoradý význam hyperglobulinémia, narušená funkcia bunkovej imunity, genetická predispozícia atď.
Patogenéza
Spomedzi početných verzií amyloidogenézy má najväčší počet priaznivcov teória dysproteinózy, lokálna bunková genéza, imunologické a mutačné teórie. Teória lokálnej bunkovej genézy berie do úvahy iba procesy prebiehajúce na bunkovej úrovni (tvorba fibrilárnych amyloidných prekurzorov makrofágovým systémom), zatiaľ čo k tvorbe a akumulácii amyloidu dochádza mimo bunky. Preto teóriu lokálnej bunkovej genézy nemožno považovať za vyčerpávajúcu.
Podľa teórie dysproteinózy je amyloid produktom abnormálneho metabolizmu proteínov. Hlavné väzby v patogenéze amyloidózy - dysproteinémia a hyperfibrinogenémia prispievajú k akumulácii hrubých proteínových a paraproteínových frakcií v plazme. Imunologická teória vzniku amyloidózy spája vznik amyloidu s reakciou antigén-protilátka, pri ktorej ako antigény pôsobia cudzie proteíny alebo produkty rozpadu vlastných tkanív. V tomto prípade dochádza k ukladaniu amyloidu najmä v miestach tvorby protilátok a nadbytku antigénov. Najuniverzálnejšia je mutačná teória amyloidózy, ktorá berie do úvahy obrovskú rozmanitosť mutagénnych faktorov, ktoré môžu spôsobiť abnormálnu syntézu bielkovín.
Amyloid je komplexný glykoproteín pozostávajúci z fibrilárnych a globulárnych proteínov úzko spojených s polysacharidmi. Amyloidné depozity sa hromadia v intime a adventícii krvných ciev, stróme parenchýmových orgánov, žľazových štruktúrach a pod. Pri miernych amyloidných depozitoch sú zmeny detekované len na mikroskopickej úrovni a nevedú k funkčným poruchám. Výrazná akumulácia amyloidu je sprevádzaná makroskopickými zmenami v postihnutom orgáne (zväčšenie objemu, mastný alebo voskový vzhľad). V dôsledku amyloidózy, stromálnej sklerózy a atrofie parenchýmu orgánov vzniká ich klinicky významný funkčný deficit.
Klasifikácia
V súlade s príčinami sa rozlišuje primárna (idiopatická), sekundárna (reaktívna, získaná), dedičná (familiárna, genetická) a senilná amyloidóza. Existujú rôzne formy dedičnej amyloidózy: stredomorská horúčka alebo periodické ochorenie (návaly horúčavy, bolesť brucha, zápcha, hnačka, zápal pohrudnice, artritída, kožné vyrážky), portugalská neuropatická amyloidóza (periférna polyneuropatia, impotencia, poruchy srdcového vedenia), fínsky typ ( atrofia rohovky, kraniálna neuropatia), dánsky variant (kardiopatická amyloidóza) a mnohé ďalšie. iní
V závislosti od prevládajúceho poškodenia orgánov a systémov nefropatické (amyloidóza obličiek), kardiopatické (amyloidóza srdca), neuropatické (amyloidóza nervového systému), hepatopatické (amyloidóza pečene), epinefropatické (amyloidóza nadobličiek ), rozlišuje sa APUD-amyloidóza, amyloidóza kože a zmiešaný typ ochorenia. Okrem toho je v medzinárodnej praxi zvykom rozlišovať medzi lokálnou a generalizovanou (systémovou) amyloidózou. Medzi lokalizované formy, ktoré sa zvyčajne vyskytujú u starších ľudí, patrí amyloidóza pri Alzheimerovej chorobe, diabetes mellitus 2. typu, endokrinné nádory, nádory kože, močového mechúra atď. V závislosti od biochemického zloženia amyloidných fibríl sa rozlišujú nasledujúce systémové formy amyloidózy :
- AL- ako súčasť fibríl, Ig ľahkých reťazcov (s Waldenströmovou chorobou, mnohopočetným myelómom, malígnymi lymfómami);
- AA- ako súčasť fibríl, sérový α-globulín akútnej fázy, svojimi vlastnosťami podobný C-reaktívnemu proteínu (pre nádorové a reumatické ochorenia, periodické ochorenia atď.);
- Ap2M- ako súčasť fibríl β2-mikroglobulínu (pri chronickom zlyhaní obličiek u pacientov na hemodialýze);
- ATTR- v zložení fibríl transportný proteín transtyretín (pri familiárnej dedičnej a senilnej forme amyloidózy).
Príznaky amyloidózy
Klinické prejavy amyloidózy sú rôznorodé a závisia od závažnosti a lokalizácie amyloidných ložísk, biochemického zloženia amyloidu, „skúsenosti“ s ochorením a od stupňa orgánovej dysfunkcie. V latentnom štádiu amyloidózy, keď amyloidné depozity možno detegovať len mikroskopicky, nie sú žiadne príznaky. S rozvojom a progresiou funkčnej nedostatočnosti jedného alebo druhého orgánu sa klinické príznaky ochorenia zvyšujú.
Pri amyloidóze obličiek je dlhodobé súčasné štádium stredne ťažkej proteinúrie nahradené rozvojom nefrotického syndrómu. Prechod do pokročilého štádia môže byť spojený s interkurentnou infekciou, očkovaním, hypotermiou, exacerbáciou základného ochorenia. Postupne sa zvyšuje edém (najskôr na nohách a potom na celom tele), vzniká nefrogénna arteriálna hypertenzia a zlyhanie obličiek. Môže sa vyskytnúť trombóza obličkových žíl. Masívna strata bielkovín je sprevádzaná hypoproteinémiou, hyperfibrinogenémiou, hyperlipidémiou a azotémiou. V moči sa zistí mikro-, niekedy makrohematúria, leukocytúria. Vo všeobecnosti sa pri amyloidóze obličiek rozlišuje skoré needematózne štádium, edematózne štádium a uremické (kachektické) štádium.
Amyloidóza srdca prebieha podľa typu reštrikčnej kardiomyopatie s typickými klinickými príznakmi - kardiomegália, arytmia, progresívne srdcové zlyhanie. Pacienti sa sťažujú na dýchavičnosť, opuch, slabosť, ktorá sa vyskytuje pri menšej fyzickej námahe. Menej často s amyloidózou srdca vzniká polyserozitída (ascites, exsudatívna pleuréza a perikarditída).
Porážka gastrointestinálneho traktu pri amyloidóze je charakterizovaná amyloidnou infiltráciou jazyka (makroglassia), pažeráka (rigidita a zhoršená peristaltika), žalúdka (pálenie záhy, nevoľnosť), čriev (zápcha, hnačka, malabsorpčný syndróm, črevná obštrukcia). Gastrointestinálne krvácanie sa môže vyskytnúť na rôznych úrovniach. S amyloidnou infiltráciou pečene sa vyvíja hepatomegália, cholestáza a portálna hypertenzia. Postihnutie pankreasu pri amyloidóze sa zvyčajne maskuje ako chronická pankreatitída.
Amyloidóza kože sa vyskytuje s výskytom viacerých voskových plakov (papuly, uzliny) na tvári, krku, prirodzených kožných záhyboch. Vonkajšie kožné lézie môžu pripomínať sklerodermiu, neurodermatitídu alebo lichen planus. Pre amyloidné lézie pohybového aparátu je typický rozvoj symetrickej polyartritídy, syndrómu karpálneho tunela, humeroskapulárnej periartrózy a myopatie. Samostatné formy amyloidózy, vyskytujúce sa pri postihnutí nervového systému, môžu byť sprevádzané polyneuropatiou, paralýzou dolných končatín, bolesťami hlavy, závratmi, ortostatickou hypotenziou, potením, demenciou atď.
Diagnostika
S klinickými prejavmi amyloidózy sa môžu stretnúť rôzni lekári: reumatológovia, urológovia, kardiológovia, gastroenterológovia, neurológovia, dermatológovia, terapeuti atď. Pre správnu diagnózu je prvoradé komplexné posúdenie klinických a anamnestických príznakov, komplexné laboratórne a inštrumentálne vyšetrenie.
Amyloidóza je systémové ochorenie, pri poruche metabolizmu bielkovín prestáva fungovať imunitný systém. V tomto ohľade sa tvorí amyloid - proteín-sacharidový komplex, ktorý sa ukladá vo všetkých tkanivách ľudských orgánov.
V priebehu času amyloid napáda orgány stále viac a viac a vytláča normálne bunky. V dôsledku toho orgán stráca svoju funkčnosť, pozorujú sa nezvratné zmeny. Ak sa ochorenie dlhodobo nelieči, dochádza k narušeniu funkcií viacerých orgánov, čo vedie k smrti.
Podľa výskumu WHO je amyloidóza diagnostikovaná asi u 1 % obyvateľov sveta. Najbežnejšia je sekundárna amyloidóza. Genetická amyloidóza je častejšie diagnostikovaná u ľudí židovskej, arménskej národnosti, ako aj u obyvateľov krajín Stredozemného mora.
Miera výskytu u mužov je dvakrát vyššia ako u žien. Medzi všetkými formami amyloidózy je diagnostikovaná nefropatická (poškodenie obličiek) a generalizovaná (poškodenie všetkých tkanív a orgánov) amyloidóza.
Typy amyloidózy, príčiny vývoja
V závislosti od príčiny amyloidózy existujú typy ochorenia, ktoré sa môžu vyvinúť nezávisle alebo v dôsledku patológií v iných systémoch a orgánoch. Vyskytujú sa tieto typy amyloidózy: senilná, s nádormi, primárna alebo idiopatická amyloidóza, dedičná, sekundárna alebo reaktívna, ako aj u pacientov podstupujúcich hemodialýzu. V závislosti od druhu prebieha vývoj amyloidózy rôzne, príznaky a prognóza sa líšia. Typy a štádiá amyloidózy budú podrobne diskutované nižšie.
Primárne (idiopatické)
Primárna amyloidóza vo väčšine prípadov začína bez dôvodu. Pri tejto forme ochorenia sa amyloid ukladá v tkanivách a orgánoch a pozoruje sa mutácia buniek imunitného systému. AL-amyloid vznikajúci pri tomto procese sa hromadí vo svaloch, koži, kardiovaskulárnom systéme a nervoch. AL-amyloid sa tiež tvorí na pozadí nádorového myelómu, keď plazmatické bunky začnú vylučovať globulíny vo veľkom objeme. Po naviazaní na plazmatické nukleoproteíny sa abnormálne globulíny premenia na amyloid.
Sekundárne (reaktívne)
Sekundárna amyloidóza sa vyvíja na pozadí progresívnych zápalových procesov v priebehu času. V tomto prípade sa tvorí AA-amyloid, ako komplikácia iných ochorení. Príčiny sekundárnej amyloidózy sú:
- Chronické infekcie - lepra, malária, tuberkulóza, syfilis, pyelonefritída, bronchiektázia.
- Hnisavé chronické ochorenia - dlhodobé hnisanie rán, osteomyelitída.
- Nádory - leukémia, lymfogranulomatóza atď.
- Prítomnosť nešpecifickej ulceróznej kolitídy (zápal hrubého čreva).
- Reumatologické ochorenie - ankylozujúca spondylitída, reumatoidná artritída atď.
Sekundárna amyloidóza môže postihnúť akýkoľvek orgán, tkanivo v tele. Zjavenie obrazu choroby nie je okamžite viditeľné. Roky po nástupe základného ochorenia je možné zaznamenať porušenie funkcií orgánu, kde sa amyloid uložil najviac. Častejšie sú touto poruchou postihnuté pečeň, obličky, slezina a lymfatické uzliny. Postupom času sú postihnuté aj iné orgány, čo vedie k zlyhaniu viacerých orgánov a smrti.
dedičná amyloidóza
Dedičná forma amyloidózy je spôsobená prítomnosťou mutovaných génov v bunkách imunitného systému. Tieto genetické mutácie sa prenášajú z generácie na generáciu, čo vedie k vytvoreniu amyloidoblastov. Dedičná forma postihuje ľudí v určitej oblasti alebo patriacich k určitej etnickej skupine. Dedičná amyloidóza sa delí na:
- Kardiopatická. Väčšinou diagnostikované u obyvateľov Dánska. Klinický obraz ochorenia pripomína primárnu amyloidózu generalizovaného typu.
- neuropatické. Je charakterizovaná poškodením nervového tkaniva. V závislosti od lokalizácie lézie sa rozlišuje portugalská (nervy nôh), americká (nervy rúk), fínska (nervový systém, očné rohovky, obličky) amyloidóza.
- Familiárne nefropatické. Ďalším názvom je anglická amyloidóza (Muckle and Wellsova choroba). Klinický obraz je urtikária, záchvaty horúčky, strata sluchu.
- Periodická (familiárna stredomorská horúčka). Choroba je bežnejšia medzi Židmi, Arabmi, Arménmi. Prejavy - teplota nad 39ºС, bolesť hlavy a svalov, nadmerné potenie. Existuje zápal membrán pľúc, peritoneálnych orgánov, synovie. Časté odchýlky v psychike.
Senilná amyloidóza
U ľudí, ktorí dosiahli vek 80 rokov, sa amyloid ukladá lokálne v rôznych tkanivách a orgánoch. Choroba je spojená s inými chorobami súvisiacimi s vekom. Existujú dva typy senilnej amyloidózy:
- Cerebrálne alebo mozgové. Vyvíja sa na pozadí Alzheimerovej choroby. Amyloid Ab sa ukladá v mozgovom tkanive.
- Srdečný. Môže ovplyvniť srdcové komory (keď sa amyloid tvorí z mutovaného krvného proteínu transtyretínu) a predsiene (keď sa amyloid tvorí z natriuretického peptidu vylučovaného srdcovými bunkami). V oboch prípadoch sa amyloidy nachádzajú v tkanivách pľúc, pankreasu a sleziny.
Pre nádory
Niektoré typy nádorov ovplyvňujú malígnu transformáciu buniek chorého orgánu, ktoré v dôsledku toho produkujú fibrilárny proteín. V tomto prípade sa amyloidóza vyvíja lokálne v tkanive orgánu postihnutého nádorom. Príčiny, ktoré vyvolávajú amyloidózu v nádoroch:
- Medulárny nádor štítnej žľazy. Rakovina sa vyvíja z C-buniek štítnej žľazy, ktoré sú normálne zodpovedné za produkciu kalcitocínu. Keď je syntéza kalcitocínu narušená, jeho fragmenty sa stávajú súčasťou amyloidu AE.
- Rakovina ostrovčekov štítnej žľazy. Ostrovčeky sú nahromadenie buniek zodpovedných za produkciu hormónov - glukagónu, inzulínu, somatostatínu atď. Malígna degenerácia buniek spôsobuje uvoľnenie fibrilárneho proteínu, ktorý následne degeneruje na amyloid.
Amyloidóza pri hemodialýze
 Hemodialýza sa nazýva život zachraňujúci postup pre pacientov, ktorých obličky nie sú schopné čistiť krv od toxínov, vedľajších produktov metabolizmu. Priraďte hemodialýzu tým, ktorí majú diagnostikované zlyhanie obličiek (akútne, chronické).
Hemodialýza sa nazýva život zachraňujúci postup pre pacientov, ktorých obličky nie sú schopné čistiť krv od toxínov, vedľajších produktov metabolizmu. Priraďte hemodialýzu tým, ktorí majú diagnostikované zlyhanie obličiek (akútne, chronické).
Podstatou zákroku je prechod krvi cez prístroj, ktorý z nej odstraňuje škodlivé látky, návrat vyčistenej krvi do tela pacienta.
Pri dialýze sa B 2 -mikroglobulín nemôže z tela vylučovať a ak je pacient dlhodobo nútený podstupovať hemodialýzu, bielkovina sa v tele hromadí v nadmernom množstve. Viaže sa na plazmatické nukleoproteíny, usadzuje sa v rôznych orgánoch a stáva sa základom amyloidu.
Príznaky amyloidózy
Vzhľadom na to, že choroba sa môže rozšíriť do akéhokoľvek orgánu alebo tkaniva, príznaky sa budú líšiť. Rôzne formy ochorenia na začiatku kurzu sú charakterizované poškodením a dysfunkciou jedného orgánu v ľudskom tele.
Postupom času choroba (ak nejde o lokálnu amyloidózu) progreduje, postihuje ďalšie orgány a tkanivá. Prejavy amyloidózy možno pozorovať v obličkách, pečeni, srdci a nadobličkách, slezine, gastrointestinálnom trakte a nervovom systéme, kĺboch, svaloch a koži. Typy ochorení sú podrobne opísané nižšie.
Poškodenie obličiek
Amyloidóza obličiek sa považuje za najnebezpečnejšiu chorobu v porovnaní s poškodením iných orgánov. Klinický obraz amyloidózy obličiek závisí od štádia. Celkovo sú ich 4 – latentné, nefrotické, azotemické, proteinurické.
V latentnom štádiu amyloidóza obličiek prakticky nevykazuje príznaky. Ak ide o sekundárnu formu, pacient pociťuje príznaky základnej choroby. Až po rokoch sa poškodenie obličiek prejaví symptomaticky.
V proteinurickom štádiu trvá amyloidóza obličiek 10 a viac rokov. V tomto čase sa amyloidóza postupne ukladá do ciev, medzibunkového priestoru a glomerulov obličiek. Z tohto dôvodu sú nefróny, ktoré zabezpečujú tvorbu moču, stlačené, atrofované a odumierajú. Integrita obličkového filtra, ktorý bežne neprepúšťa veľké molekulárne proteíny a krvinky, je narušená. Následne sa bielkoviny vylučujú močom. V tomto štádiu je ťažké mať podozrenie na amyloidózu obličiek, pretože vylučovacia funkcia nie je narušená. Problém nájdete vo výsledkoch laboratórnych testov.
Renálna amyloidóza v nefrotickom štádiu sa prejavuje ďalšou deštrukciou obličkového filtra. Z tohto dôvodu sa veľké množstvo bielkovín stráca močom, ich koncentrácia v krvi klesá. Proteíny sú súčasťou procesu udržiavania krvi v cievach. S poklesom koncentrácie bielkovín vstupuje kvapalina do tkanív, opuch sa vyskytuje kedykoľvek počas dňa, bez ohľadu na polohu tela. Ďalej postupuje amyloidóza obličiek, edém je silne výrazný. Tekutina sa hromadí v pobrušnici, pleurálnej dutine, srdcovom vaku. Toto štádium trvá 4-6 rokov.
V azotemickom štádiu funguje iba 25 % celkového objemu obličkového tkaniva. To nestačí na odstránenie škodlivých toxínov, močoviny, a preto ich koncentrácia rastie. Klinický obraz zlyhania obličiek sa prejavuje nasledovne:
- zhoršené močenie. Namiesto predpísaných 800 ml denne pacient vylúči menej ako 50 ml moču;
- zdravie sa zhoršuje, objavuje sa slabosť, únava;
- trávenie je narušené, chuť do jedla zmizne, objavuje sa nevoľnosť a vracanie, sucho v ústach je sprevádzané nepríjemným zápachom;
- koža sa stáva bledá, suchá, neustále svrbivá;
- kardiovaskulárny systém trpí, čo spôsobuje arytmiu, krvný tlak stúpa, je možný nárast srdcového svalu;
- mozog pod vplyvom vysokej koncentrácie kyseliny močovej je poškodený, objavuje sa nespavosť a poruchy pamäti, podráždenosť, pokles duševných schopností;
- zníženie hemoglobínu a červených krviniek vedie k anémii.
Poškodenie pečene
Systémová amyloidóza sa často prejavuje poškodením pečene. Amyloidné usadeniny vyvíjajú tlak na žlčové cesty, krvné cievy a pečeňové bunky, čo vedie k narušeniu funkcie orgánov. Zvýraznenie syndrómov amyloidózy naznačuje zvýšenie pečene, pociťované pri palpácii.
Povrch pečene zostáva hladký, nie je žiadna bolesť. Pri dlhom priebehu ochorenia sa zriedkavo vyvinie zlyhanie pečene, ktoré je spojené s regeneračnými schopnosťami orgánu.
Amyloidóza pečene sa prejavuje nasledujúcimi príznakmi:
- Zväčšenie pečene.
- portálna hypertenzia. Normálne sa krv z vnútorných orgánov dostáva do pečene, kde sa čistí a potom sa vracia do krvného obehu. Pri stláčaní ciev pečene amyloidom sa zvyšuje tlak v žilách vnútorných orgánov. V dôsledku toho sú opuchy nôh, hnačka s krvou, krvácanie v gastrointestinálnom trakte.
- Žltačka je zriedkavá, iba v prípade kompresie žlčových ciest amyloidnými ložiskami. Ak je to dôvod, svrbenie bude sprevádzať žltačku.
Zástava srdca
Amyloidóza srdca sa vyvíja v primárnych a iných formách dedičnej povahy. V dôsledku usadenín amyloidu v myokarde a membránach srdca je narušený krvný obeh, svalové bunky odumierajú.
Príznaky ochorenia:
- arytmia;
- reštriktívna kardiomyopatia;
- zástava srdca.
Arytmia sa vyskytuje na pozadí amyloidných usadenín v srdcovom svale, čo narúša vedenie nervového impulzu. V dôsledku toho sa komory srdca sťahujú nerovnomerne, objavuje sa arytmia. Pacient pociťuje závraty, pozoruje sa mdloby. V dôsledku zhoršeného prívodu krvi do mozgu je možný smrteľný výsledok.
Reštriktívna kardiomyopatia sa vyskytuje na pozadí amyloidných usadenín v myokarde. V dôsledku toho sa srdcový sval zahusťuje, stáva sa menej roztiahnuteľným, čo vedie k zlému fungovaniu srdcových komôr. Klinickým obrazom ochorenia je únava, dýchavičnosť, prudký pokles krvného tlaku pri zmene z vodorovnej polohy do zvislej.
Pri zlyhaní srdca je narušený krvný obeh v tele. To sa prejavuje opuchom, dýchavičnosťou. Srdcové zlyhanie pri amyloidóze nie je prístupné štandardnej liečbe kardiovaskulárnych ochorení. Choroba postupuje rýchlo, čo vedie k smrti v priebehu niekoľkých mesiacov.
Poškodenie nadobličiek a sleziny
 Nadobličky sú žľazy umiestnené na každej obličke a sú zodpovedné za vylučovanie hormónov. Amyloidóza narúša funkciu orgánov zastavením syntézy hormónov. Ak je amyloid uložený v slezine, orgán sa zväčšuje, čo je viditeľné pri palpácii.
Nadobličky sú žľazy umiestnené na každej obličke a sú zodpovedné za vylučovanie hormónov. Amyloidóza narúša funkciu orgánov zastavením syntézy hormónov. Ak je amyloid uložený v slezine, orgán sa zväčšuje, čo je viditeľné pri palpácii.
Normálne slezina vylučuje z krvného obehu deformované bunky, ktoré uviazli v jej štruktúre. Amyloidné usadeniny v slezine spôsobujú uviaznutie zdravých červených krviniek, krvných doštičiek a bielych krviniek.
V dôsledku toho vzniká anémia (celková slabosť, bledosť kože, dýchavičnosť), trombocytopénia (krvácanie z nosa, kožné krvácanie), leukopénia (náchylnosť na infekcie).
Gastrointestinálna lézia
Črevná amyloidóza môže byť generalizovaná, keď je narušená absorpcia živín, a lokálna, keď akumulácia amyloidu napodobňuje nádor. V prvom prípade sa objavia príznaky ako hnačka, chudnutie, slabosť, duševné poruchy, anémia. V druhom prípade je ochorenie charakterizované zápchou, bolesťou brucha, nadúvaním.
Poškodenie kĺbov a svalov
Amyloid postihuje najskôr drobné kĺby na chodidlách, rukách, ako choroba postupuje, usadzuje sa v lakťoch a kolenách. Ochorenie je charakterizované bolesťou pri pohybe, opuchom tkaniva a začervenaním kože, horúčkou v postihnutej oblasti, poruchou funkcie kĺbu.
Amyloid sa dlhodobo nenápadne ukladá v spojivovom tkanive, bez narušenia svalovej štruktúry a bez toho, aby sa objavil. V priebehu času sú bunky svalového tkaniva stlačené, prívod krvi do nich je narušený a zomierajú. Ochorenie je charakterizované svalovou slabosťou, bolesťou, stuhnutím a svalovou hypertrofiou.
Diagnóza amyloidózy
Na diagnózu, akou je amyloidóza, môžu mať podozrenie lekári rôznych špecializácií – reumatológovia, kardiológovia a urológovia, neurológovia, dermatológovia a pod. Preto by mala byť diagnóza amyloidózy založená na komplexnom zhodnotení anamnézy, klinických príznakov, laboratórneho a inštrumentálneho vyšetrenia. . Na vyšetrenie stavu orgánov je predpísané EKG, röntgenové vyšetrenie pažeráka, endoskopia, sigmoidoskopia. Ak existuje podozrenie na amyloidózu obličiek, diagnóza nevyhnutne zahŕňa ultrazvuk brušnej dutiny.
Liečba amyloidózy
Napriek tomu, že existujú rôzne závažné ochorenia, amyloidóza má zlú prognózu. Faktom je, že v počiatočných štádiách nie je možné ochorenie identifikovať a jeho klinické prejavy sú viditeľné mnoho rokov po nástupe ochorenia. Pri takejto diagnóze, ako je amyloidóza obličiek, je liečba iba podporná, pretože terapeutické opatrenia nie sú účinné.
Pri prvom podozrení na prítomnosť ochorenia je nutná hospitalizácia na nefrológii na vyšetrenie urogenitálneho systému, keďže za najnebezpečnejší prejav sa považuje poškodenie obličiek. Na vyšetrenie prítomnosti poškodenia iných orgánov sú zapojení aj ďalší špecialisti.
Ak diagnóza neodhalila vážne porušenia vo fungovaní životne dôležitých orgánov, liečba amyloidózy sa môže vykonávať doma, kde pacient musí prísne dodržiavať všetky predpisy lekára. Liečba môže zahŕňať lieky, diétu, dialýzu a transplantáciu orgánov.
"Amyloidóza" je termín, ktorý spája skupinu ochorení, ktoré sa vyznačujú širokou škálou klinických prejavov a sú charakterizované extracelulárnym ukladaním nerozpustných patologických fibrilárnych proteínov v orgánoch a tkanivách. Táto patológia bola prvýkrát opísaná v 17. storočí. Bonnet - ságová slezina u pacienta s pečeňovým abscesom. V polovici XIX storočia. Virchow použil botanický výraz „amyloid“ (z gréckeho amylon, škrob) na opis extracelulárneho materiálu nájdeného v pečeni pri pitve, pretože veril, že má podobnú štruktúru ako škrob. Následne sa stanovila proteínová povaha ložísk, no pojem „amyloid“ sa zachoval dodnes.
V 20. rokoch. V 20. storočí Benhold navrhol zafarbiť amyloid konžskou červenou, potom bol objavený efekt dvojitého lomu v polarizovanom svetle – zmena z tehlovočervenej na jablkovo zelenú. V roku 1959 Cohen a Calkins stanovili fibrilárnu štruktúru amyloidu pomocou elektrónovej mikroskopie.
Klinické koncepty amyloidózy tiež prešli evolúciou: Rokitansky v roku 1842 vytvoril spojenie medzi „mazovou chorobou“ a tuberkulózou, syfilisom a rickettsiózou; Wilks v roku 1856 opísal „tukové orgány“ u pacienta, ktorý nemal žiadne sprievodné ochorenia; Atkinson v roku 1937 objavil amyloidózu u pacientov s mnohopočetným myelómom. Bola identifikovaná senilná (Soika, 1876) a dedičná (Andrade, 1952) forma ochorenia, amyloidóza bola rozdelená na genetickú, primárnu a sekundárnu a nakoniec bola v roku 1993 prijatá klasifikácia WHO na základe špecifickosti hlavný fibrilárny amyloidový proteín.
V našej krajine výrazne prispeli k rozvoju myšlienok o amyloidóze E. M. Tareev, I. E. Tareeva, V. V. Serov. Obrovská úloha pri štúdiu primárnych a genetických variantov amyloidózy a periodických ochorení patrí O. M. Vinogradovej, ktorej monografie vydané v rokoch 1973 a 1980 dnes nestratili svoj význam.
V súčasnosti sa amyloidóza klinicky delí na systémové a lokálne formy. Medzi systémovými formami, v závislosti od zloženia fibrilárnych ložísk, existujú štyri typy ( ).
Medzi lokálne formy amyloidózy v súčasnosti patrí Alzheimerova choroba (A-beta, fibrily pozostávajú z β-proteínu uloženého v mozgu), amyloidóza pankreatických ostrovčekov, ktorá môže mať patogenetický vzťah s diabetom 2. typu, amyloidóza, ktorá sa vyskytuje pri endokrinných nádoroch, amyloidné nádory kože, oblasti nosohltanu, močového mechúra a iných zriedkavých typov.
AL amyloidóza
Rozvoj AL-amyloidózy je možný pri mnohopočetnom myelóme, Waldenströmovej chorobe, B-bunkových lymfómoch a pri primárnej amyloidóze môže byť idiopatický. Všetky tieto varianty spája spoločná patogenéza, primárnu amyloidózu je najťažšie rozpoznať kvôli absencii zjavných príznakov hematologického ochorenia, preto stojí za to podrobne sa zaoberať touto formou.
Pri primárnej amyloidóze, benígnej dyskrázii plazmatických buniek súvisiacej s mnohopočetným myelómom, abnormálne klony plazmatických buniek kostnej drene produkujú amyloidogénne imunoglobulíny. Niektoré aminokyseliny vo variabilných oblastiach ľahkých reťazcov týchto imunoglobulínov zaujímajú nezvyčajnú polohu, čo vedie k ich nestabilite a spôsobuje sklon k fibrilogenéze. U pacientov s primárnou amyloidózou je obsah plazmatických buniek v kostnej dreni zvýšený na 5-10% (normálne menej ako 4%, s mnohopočetným myelómom - viac ako 12%) a produkujú určitý izotyp ľahkých reťazcov imunoglobulínu, ktorý prevláda pri imunohistochemickom farbení. Voľné monoklonálne ľahké reťazce prevládajúceho izotypu lambda alebo (menej často) kappa sa zisťujú v krvi a moči, ale ich obsah je nižší ako pri mnohopočetnom myelóme.
Klinický obraz primárnej amyloidózy je rôznorodý a je determinovaný prevažujúcim zapojením niektorých orgánov do patologického procesu – srdca, obličiek, nervového systému, gastrointestinálneho traktu, pečene a pod. Prvými príznakmi sú slabosť a strata hmotnosti, ale pri tomto štádiu, pred objavením sa orgánových symptómov , je diagnóza extrémne zriedkavá.
Najčastejšími cieľovými orgánmi pri AL amyloidóze sú obličky a srdce. Poškodenie obličiek sa prejavuje nefrotickým syndrómom, pretrvávajúce a s nástupom chronického zlyhania obličiek nie sú typické hematúria a artériová hypertenzia.
S ukladaním amyloidu v myokarde vznikajú rôzne poruchy rytmu, progresívne srdcové zlyhanie, ktorému môžu predchádzať asymptomatické zmeny na EKG v podobe poklesu napätia zubov. Echokardiografia odhaľuje koncentrické zhrubnutie stien ľavej a pravej komory, zmenšenie objemu srdcových dutín, mierny pokles ejekčnej frakcie a diastolickú dysfunkciu myokardu ľavej komory.
Často sú príznaky postihnutia nervového systému - autonómne, vo forme ortostatickej hypotenzie a periférne - vo forme porúch citlivosti. V posledných rokoch boli opísané aj lézie centrálneho nervového systému, hoci predtým sa verilo, že nie sú charakteristické pre primárnu amyloidózu.
Dyspeptické javy (pocit plnosti, zápcha, hnačka) a malabsorpčný syndróm môžu byť spôsobené tak poškodením autonómneho nervového systému, ako aj amyloidózou gastrointestinálneho traktu. Veľmi charakteristická je hepatomegália, ktorej charakter by sa mal rozlišovať medzi preťažením v dôsledku zlyhania srdca a amyloidným poškodením pečene. To je potvrdené zvýšením hladiny alkalickej fosfatázy v krvnom sére. Často je postihnutá slezina, ale nie vždy sa zistí splenomegália a nemá veľký klinický význam.
Makroglossia, klasický znak primárnej amyloidózy, sa vyskytuje u 20 % pacientov, infiltrácia mäkkých tkanív môže viesť k svalovej a kožnej atrofii, dystrofii nechtov, alopécii a vzniku nádorových útvarov – amyloidu.
Menej časté je poškodenie ciev, ktorého príznakmi sú periorbitálna purpura – „mývalie oči“ a ekchymóza. Môže sa vyskytnúť krvácanie, vrátane krvácania z močového mechúra, spôsobené zmenami v cievnej stene a porušením koagulačného systému, predovšetkým nedostatkom faktora X, ktorý sa viaže na amyloid. Je obvyklé vysvetliť trombocytózu charakteristickú pre amyloidózu nedostatkom koagulačných faktorov.
Pľúcna amyloidóza sa často zistí iba pri pitve. V niektorých prípadoch však môže byť dýchavičnosť, hemoptýza a hydrotorax spôsobené nielen kongestívnym srdcovým zlyhaním a nefrotickým syndrómom, ale aj amyloidným ochorením pľúc. Je možné ukladanie amyloidu v alveolách a rozvoj pľúcneho amyloidómu. Rádiograficky možno zistiť sieťové a nodulárne zmeny v pľúcnom tkanive.
Postihnutie nadobličiek môže viesť k nedostatočnosti nadobličiek, ktorá sa často nerozpozná, pretože hypotenzia a hyponatriémia sa považujú za symptómy zlyhania srdca a poškodenia autonómneho nervového systému. U 10-20% pacientov sa môže vyskytnúť hypotyreóza ako prejav poškodenia štítnej žľazy, často dochádza k zväčšeniu podčeľustných slinných žliaz.
Diagnóza primárnej amyloidózy, okrem indikovaných klinických príznakov, ktoré môžu byť podobné pri sekundárnej amyloidóze, je založená na množstve laboratórnych údajov. U 85 % pacientov imunoelektroforéza proteínov krvného séra a moču odhalí monoklonálne imunoglobulíny. V rutinných štúdiách sa rovnaké monoklonálne imunoglobulíny nachádzajú v moči vo forme proteínu Bence-Jones. Biopsia kostnej drene umožňuje diferenciálnu diagnostiku mnohopočetného myelómu, ako aj detekciu mierneho zvýšenia počtu plazmatických buniek a ich monoklonality imunohistochemickým farbením.
Avšak ani kombinácia charakteristického klinického obrazu a prítomnosti monoklonálnych plazmatických buniek a proteínov je stále nedostatočná na potvrdenie diagnózy primárnej amyloidózy. Rozhodujúcu úlohu tu zohrávajú údaje z biopsie. Najmenej invazívna je aspirácia podkožného tukového tkaniva prednej brušnej steny, ktorá dáva 80-90% pozitívnych výsledkov pri AL-amyloidóze (u nás sa táto metóda zatiaľ nepoužíva). Biopsia ďasien a rektálnej sliznice má určitú diagnostickú hodnotu, ale percento pozitívnych výsledkov sa značne líši v závislosti od štádia procesu, preto je vhodné vykonať biopsiu jedného z postihnutých orgánov – obličky, pečeň , srdce, čo dáva takmer 100% pozitívne výsledky pri amyloidóze AL typu.
V prvom rade sa bioptický materiál zafarbí konžskou červenou. Ak sa zistí kongofília skúmaného materiálu, je potrebné ju študovať v polarizovanom svetle, efekt dvojlomu je charakteristický len pre amyloid, ostatné kongofilné látky nezískajú jablkovo zelenú farbu. Potom je žiaduce typizácia amyloidu. Najpresnejšia je imunohistochemická metóda využívajúca monoklonálne protilátky proti amyloidovým prekurzorovým proteínom. V súčasnosti je však u nás prakticky nedostupný. Preto sa na diagnostiku používa farbenie roztokmi alkalického guanidínu alebo manganistanu draselného, čo umožňuje, aj keď nepriamo, určiť typ fibrilárnych ložísk.
Prognóza primárnej amyloidózy je horšia ako u iných foriem ochorenia, priemerná dĺžka života nepresahuje dva roky, v prítomnosti srdcových ochorení alebo multisystémových lézií bez liečby pacienti zomierajú v priebehu niekoľkých mesiacov. Najčastejšími príčinami smrti sú zlyhanie srdca a obličiek, sepsa, cievne komplikácie a kachexia. Patogenetická podobnosť s mnohopočetným myelómom nám umožňuje počítať s inhibíciou progresie ochorenia počas chemoterapie, ktorá sa vykonáva na potlačenie monoklonálnych plazmatických buniek. Existuje niekoľko liečebných režimov ().
Použitie chemoterapie v prípade úspešnej liečby môže zvýšiť dĺžku života pacientov na obdobie 10 až 18 mesiacov. Účinnosť liečby je však nízka, najmä v dôsledku skutočnosti, že v mnohých prípadoch vedie progresia ochorenia k úmrtiu pacientov pred ukončením liečby, ako aj v dôsledku rozvoja cytopénie, infekčných chorôb. komplikácie, fatálne arytmie pri liečbe ultravysokými dávkami dexazónu. Použitie vysokých dávok melfolanu s transplantáciou autológnych kmeňových buniek umožňuje dosiahnuť remisiu vo viac ako 50% prípadov, avšak použitie tejto metódy je limitované závažnosťou stavu, vekom pacientov a funkčnými poruchami srdce a obličky. V mnohých prípadoch je možná len symptomatická podporná liečba.
AA amyloidóza
K rozvoju AA-amyloidózy dochádza pri chronických zápalových procesoch, prekurzormi AA-amyloidu sú sérové proteíny akútnej fázy, α-globulíny produkované bunkami rôznych typov, hlavne neutrofilmi a fibroblastmi. Sekundárna amyloidóza sa vyvíja pri reumatoidnej artritíde, Bechterevovej chorobe, psoriatickej artritíde, rôznych nádoroch, Hodgkinovej chorobe, ulceróznej kolitíde a Crohnovej chorobe, periodickom ochorení (familiárna stredomorská horúčka), ako aj tuberkulóze, osteomyelitíde, bronchiektázii.
Charakteristickými klinickými znakmi AA amyloidózy sú u väčšiny pacientov poškodenie obličiek, ako aj relatívne zriedkavé poškodenie pečene a/alebo sleziny (asi 10 %) a srdca (detegované iba echokardiografiou). Makroglossia nie je typická pre sekundárnu amyloidózu. Diagnóza je založená na kombinácii renálnej amyloidózy a chronického zápalového ochorenia, čo sa potvrdí imunohistochemickým farbením bioptického materiálu, u nás sa používajú už vyššie uvedené nepriame metódy farbenia.
Prognóza do značnej miery závisí od charakteru základného ochorenia, pri prirodzenom priebehu sa u tretiny pacientov po 5 rokoch od zistenia proteinúrie rozvinie zlyhanie obličiek. Pri periodickom ochorení je päťročná miera prežitia 25%.
Liečba je založená na potlačení ohniska - zdroja produkcie sérových prekurzorových proteínov. Odstránenie nádorov, sekvestrektómia, resekcia čreva, liečba tuberkulózy, zníženie aktivity reumatoidnej artritídy (s použitím cytostatík) vedú k zastaveniu progresie amyloidózy, niekedy k opačnému rozvoju klinických prejavov, najmä nefrotických syndróm.
Použitie kolchicínu pri periodickom ochorení je metódou voľby, jeho účinnosť je preukázaná, liečba zabraňuje vzniku amyloidózy a spomaľuje jej progresiu. Pri iných formách sekundárnej amyloidózy sa účinnosť kolchicínu nepotvrdila.
Senilné a dedičné formy systémovej amyloidózy, ako aj lokálne formy sú zriedkavé, dialyzačná amyloidóza je odborníkom dobre známa, v bežnej praxi sa s ňou takmer nestretávame.
Symptomatická liečba nezávisí od typu amyloidózy, ale od postihnutých cieľových orgánov ( ).
Amyloidóza, najmä primárna, sa považuje za zriedkavú patológiu, ale v skutočnosti nie je taká zriedkavá, ako je ťažké ju diagnostikovať. Adekvátna diagnóza si vyžaduje nielen znalosť kliniky a patogenézy tohto ochorenia, ale aj dostupnosť určitých diagnostických schopností. Na ilustráciu tohto bodu uvádzame naše vlastné údaje ( ). Na nefrologickom oddelení Moskovskej mestskej klinickej nemocnice pomenovanej po S.P. Botkinovi v rokoch 1993-2003. Bolo pozorovaných 88 pacientov, u ktorých bola diagnostikovaná amyloidóza.
Diagnóza bola morfologicky potvrdená u všetkých pacientov s AL-amyloidózou, senilnou a nešpecifikovanou amyloidózou a u 30 pacientov so sekundárnou amyloidózou – spolu 53 prípadov. Biopsia obličky bola vykonaná u 12 pacientov, biopsia pečene bola vykonaná u 2 pacientov, biopsia čreva bola vykonaná u 8 pacientov, ďasná boli vykonané v 12 prípadoch a v 19 prípadoch bola diagnóza potvrdená morfologickým vyšetrením rezového materiálu.
Vo väčšine prípadov bola diagnóza amyloidózy prvýkrát stanovená ako výsledok vyšetrenia na nefrologickom oddelení. Porovnali sme odporúčacie a klinické diagnózy u pacientov s AL-amyloidózou ( ).
Len v dvoch prípadoch z 20 (10 %) bola odporúčaná diagnóza „primárna amyloidóza“, pričom u jedného z týchto pacientov bola stanovená na Klinike MMA pre liečbu a choroby z povolania, u druhého na zahraničnej klinike.
Všetci pacienti, u ktorých bol diagnostikovaný mnohopočetný myelóm s rozvojom AL-amyloidózy, boli preložení na hematologické oddelenia. Z 11 pacientov s primárnou amyloidózou dostávalo sedem pacientov intermitentnú chemoterapiu s kombináciou melfolanu a perorálneho prednizolónu, štyria z nich v kombinácii s dialyzačnou liečbou a ďalší pacient dostával iba dialýzu a symptomatickú liečbu. Z týchto pacientov päť zomrelo v priebehu dvoch týždňov až dvoch rokov od začiatku liečby (všetci so zlyhaním obličiek a poškodením viacerých orgánov), jeden pacient je na dialýze, jeden pacient bol odoslaný na autológnu transplantáciu kmeňových buniek a jeden pacient dostáva liečbu až do prítomného času. U jedného pacienta bola chemoterapia oneskorená pre prítomnosť dlhodobého nezjazvujúceho žalúdočného vredu a ďalší dvaja pacienti liečbu odmietli.
Medzi pacientmi so sekundárnou amyloidózou v našej štúdii prevládali pacienti s reumatoidnou artritídou, na druhom mieste medzi príčinami boli chronická osteomyelitída a psoriatická artritída, ostatné ochorenia boli menej časté ( ).
Liečba reumatoidnej artritídy a psoriatickej artritídy prebiehala s použitím cytostatík (metatrexát, azatioprín), aj keď v mnohých prípadoch boli možnosti terapie obmedzené pre prítomnosť CRF a komorbidít. Pacienti s chronickou osteomyelitídou boli odoslaní na oddelenia purulentnej chirurgie. Pacienti s Bechterevovou chorobou a Crohnovou chorobou dostávali špecifickú liečbu, pacienti s CHOCHP a tuberkulózou boli odosielaní aj do špecializovaných nemocníc. Jeden z pacientov s nádorom žalúdka bol úspešne operovaný a v priebehu štyroch rokov pozorovania došlo k postupnej regresii nefrotického syndrómu, v ostatných prípadoch nádorov prevalencia procesu umožňovala len symptomatickú terapiu, pacient s lymfogranulomatóza bola prijatá v terminálnom stave. Úmrtnosť medzi pacientmi so sekundárnou amyloidózou bola 38 % (v dôsledku pacientov s pokročilými léziami v čase diagnózy). Všetci pacienti s periodickým ochorením dostávali terapiu kolchicínom.
Charakteristiky diagnostiky a aplikácie moderných metód liečby primárnej amyloidózy možno ilustrovať na nasledujúcom príklade: pacientka K., 46-ročná, bola prvýkrát hospitalizovaná koncom októbra 2002 so sťažnosťami na opuchy nôh, búšenie srdca, amenoreu. Má v anamnéze prechladnutie, apendektómiu, dva normálne urgentné pôrody, príznaky ochorenia obličiek a nemá žiadne chronické ochorenia. V apríli 2002 prekonala akútny zápal pľúc v hornom laloku pravých pľúc, bola liečená ambulantne, dostávala injekcie abaktalu a linkomycínu. Pre lokalizáciu zápalu pľúc bola vyšetrená v tuberkulóznej ambulancii, diagnóza tuberkulóza bola vylúčená. Začiatkom júna sa prvýkrát objavili edémy na nohách, na ktoré nebola vyšetrená. Edém po krátkom čase nezávisle eliminovaný, potom obnovený. Pacient bol hospitalizovaný v terapeutickej nemocnici, vyšetrením bola zistená proteinúria do 1,65 %, hypoproteinémia (celkový sérový proteín 52 g/l), krvný tlak v norme (120/80 mm Hg), močový sediment nezmenený, plazmatické hladiny kreatinínu boli tiež v normálnych medziach. Bola stanovená diagnóza "akútna glomerulonefritída", bola vykonaná liečba ampicilínom, zvonkohrou, heparínom, triampurom, bola vykonaná tonzilektómia. Proteinúria pretrvávala, edém sa postupne zvyšoval, a preto bol pacient s diagnózou chronická glomerulonefritída odoslaný do nemocnice na ďalšie vyšetrenie a liečbu. S. P. Botkin.
Pri vyšetrení je koža čistá, normálnej farby, anasarka, opuch je masívny, hustý, ascites je určený, periférne lymfatické uzliny nie sú zväčšené. TK 110/70 mm Hg. Art., ozvy srdca sú zvukové, čisté, rytmické, tep 90 úderov/min., pečeň a slezina nie sú zväčšené, diuréza do 1000 ml/deň, stolica pravidelná, bez patologických nečistôt. Vyšetrením bol zistený nefrotický syndróm - proteinúria 3 g/l, močový sediment malý, hypodysproteinémia, hyperlipidémia (celkový sérový proteín 39 g/l, albumín 12 g/l, globulíny 7-30-15-19 %, resp. α 1 -α 2 -β-γ cholesterol 17,8 mmol/l, β-lipoproteíny 250 U), pri analýze moču na Bence-Jonesov proteín je reakcia negatívna, denné vylučovanie 17-KS nie je znížené. Klinický krvný test a ostatné biochemické parametre sú v norme, koagulogram - ťažká hyperfibrinogenémia, zvýšená hladina RKFM. Štúdium krvných imunoglobulínov: Ig-A - 0,35, Ig-M - 35,7 (dve normy), Ig-G - 1,96 g / l. RTG hrudníka, kostí lebky a panvy, ultrazvuk dutiny brušnej, obličiek, štítnej žľazy, ECHO-KG bez patológie, ultrazvuk malej panvy - príznaky adenomyózy tela maternice, endoskopia - refluxná ezofagitída, chronická gastritída. Pri vyšetrení neuropatológom nebola zistená žiadna patológia, onkológ diagnostikoval fibrocystickú mastopatiu.
Pre objasnenie genézy nefrotického syndrómu v lokálnej anestézii pod ultrazvukovým vedením bola vykonaná tenkoihlová punkčná biopsia pravej obličky, bez komplikácií. Pri štúdiu biopsie v mezangiu glomerulov a v extraglomerulárnych cievach sa zaznamenáva ukladanie amyloidu. Amyloid zaťažuje až 25 % glomerulárnych cievnych slučiek. Imunohistochemická štúdia neodhalila špecifickú luminiscenciu. Pri ošetrení prípravkov alkalickým roztokom guanidínu počas 2 hodín sa zachovajú kongofília a ich vlastnosti v polarizovanom svetle, čo je typické pre AL-amyloidózu.
Na objasnenie povahy AL-amyloidózy sa v laboratóriu Immunotest uskutočnila imunochemická štúdia krvi a moču. M-lambda paraproteinémia bola odhalená s poklesom hladiny polyklonálnych imunoglobulínov a paraproteinúriou Bence-Jonesovho typu lambda na pozadí masívnej neselektívnej proteinúrie. Pacient bol konzultovaný hematológom, bola prítomná Waldenströmova choroba a bola vykonaná trepanobiopsia kostnej drene. Záver: v existujúcich dutinách kostnej drene sú viditeľné bunky všetkých troch klíčkov normálnej hematopoézy, ako aj lymfoidné bunky, ktoré netvoria zhluky. Diagnóza Waldenströmovej choroby bola zamietnutá z dôvodu absencie lymfoidnej infiltrácie kostnej drene, zväčšených lymfatických uzlín a sleziny a absencie nádorového substrátu.
Stanovená diagnóza primárna amyloidóza s poškodením obličiek, nefrotický syndróm, zachovaná funkcia obličiek, známky iného orgánového poškodenia neboli zistené. Od januára 2003 sa začala chemoterapia s melfolánom 16 mg/deň a prednizolónom 100 mg/deň, štyri dni každých šesť týždňov. Vykonáva sa aj symptomatická liečba: furosemid, veroshpiron, prípravky draslíka, famotidín, transfúzie albumínu. Doteraz bolo vykonaných päť cyklov chemoterapie s dobrou toleranciou, edémy sa znížili, proteinúria klesla na 1,8 g/l, závažnosť hypodysproteinémie sa mierne znížila (celkové bielkoviny 46 g/l, albumíny 18 g/l, α 2 -globulíny 20%). Funkcia obličiek zostáva zachovaná, plazmatický kreatinín je 1,3 mg/dl, pri kontrolných dynamických vyšetreniach neboli zistené žiadne známky poškodenia iných orgánov a systémov.
Tento prípad názorne ilustruje skutočnosť, že na diagnostiku amyloidózy sú potrebné morfologické, imunologické a imunochemické vyšetrenia. U nášho pacienta bola teda najzrejmejšou klinickou diagnózou „chronická glomerulonefritída“ a pri absencii možnosti vykonania biopsie obličky by táto diagnóza bola s najväčšou pravdepodobnosťou stanovená. Pacient nemal žiadne klinické indikácie systémového charakteru ochorenia, chronického zápalového procesu, ochorenia krvného systému, s výnimkou zvýšenia hladiny Ig-M. A iba údaje získané v štúdii biopsie obličiek viedli k trepanobiopsii kostnej drene a imunochemickej štúdii, ktorá spolu umožnila diagnostikovať primárnu amyloidózu pred objavením sa systémového poškodenia. Patogenetická terapia bola zahájená síce na pozadí už rozvinutého nefrotického syndrómu, ale pred vznikom renálneho zlyhania a len s 25 % glomerulov zaťažených amyloidom, čo je prognosticky relatívne priaznivé.
Záverom konštatujeme, že amyloidóza je závažné ochorenie s vysokou úmrtnosťou, ktoré je mimoriadne ťažké diagnostikovať, avšak včasné a kvalitné vyšetrenie pacientov umožňuje včasnejšiu diagnostiku a včasné podanie adekvátnej terapie v r. obrat, umožňuje zlepšiť prognózu v tomto období.skupina pacientov.
Literatúra
- Varshavsky V. A., Proskurneva E. P. Význam a metódy morfologickej diagnostiky amyloidózy v modernej medicíne // Praktická nefrológia. - 1998. - 2:16-23.
- Vinogradova OM Primárne a genetické varianty amyloidózy. — M.: Medicína, 1980.
- Zakharova E. V., Khrykina A. V., Proskurneva E. P., Varshavsky V. A. Prípad primárnej amyloidózy: ťažkosti s diagnostikou a liečbou // Nefrológia a dialýza. - 2002. - 1:54-61.
- Rameev VV Zvláštnosti poškodenia obličiek pri AA a AL-amyloidóze: diss... cand. med. vedy. - M., 2003.
- Kozlovskaya L. V., Varshavsky V. A., Chegaeva T. V. a kol. Amyloidóza: moderný pohľad na problém // Praktická nefrológia. - 1998. - 2:24-26.
- Rodney H., Raymond L.C a Skinner M. // The systemic Amyloidoses; New England Journal of Medicine, 1997. 337:898-909.
- Dhodapkar M.V., Jagannath S., Vesole d. et al // Liečba AL-amyloidózy dexametazónom plus interferón alfa / Leuc lymfóm. 1997. - 27 (3-4): 351-365
- Gertz M.A., Lacy M.Q., Lust J.A. et all // Štúdia fázy II s vysokou dávkou dexametazónu pre predtým liečenú amyloidózu ľahkého reťazca imunoglobulínu. Am J Hematol, 1999, 61 (2): 115-119.
- Gertz M.A., Lacy M., Q., Lust J.A. et al // Štúdia fázy II s vysokou dávkou dexametazónu u neliečených pacientov s primárnou systémovou amyloidózou. Med Oncol 1999.-16(2):104-109
- Sezer O., Schmid P., Shweigert M. et al // Rýchla reverzia nefrotického syndrómu v dôsledku primárnej systémovej AL amyloidózy po VAD a následnej vysokodávkovej chemoterapii s podporou autológneho kmeňa. Transplantácia kostnej drene. 1999. - 23(9): 967-969.
- Sezer O., Neimoller K., Jakob C. et al // Nové prístupy k liečbe primárnej amyloidózy. Expert Opin Investig Grugs. 2000. - 9(10):2343-2350
- Sezer O., Eucker J., Jakob C., Possinger K. // Diagnostika a liečba AL amyloidózy. Clinic Nephrol. 2000.-53(6):417-423.
- Skinner M. "Amyloidóza" Súčasná terapia v alergii, imunológii a reumatológii. Mosby-Year Book. 1996. - 235-240.
- Palladini G., Anesi E., Perfetti V. a kol. Modifikovaný režim vysokých dávok dexametazónu pre primárnu systémovú (AL) amyloidózu. British Journal of Hematology. 2001.-113:1044-1046.
E. V. Zacharovej
Moskovská mestská klinická nemocnica. S. P. Botkina
Tabuľka 2. Liečebné režimy pre primárnu amyloidózu
- Cyklické perorálne podávanie melfolánu (0,15 – 0,25 mg/kg telesnej hmotnosti za deň) a prednizolónu (1,5 – 2,0 mg/kg denne) počas štyroch až siedmich dní každé štyri až šesť týždňov počas roka, až do dosiahnutia dávky v priebehu 600 mg
- Perorálne užívanie melfolánu v dávke 4 mg / deň počas troch týždňov, potom po dvojtýždňovej prestávke - 2-4 mg / deň štyri dni v týždni nepretržite, až kým sa nedosiahne dávka 600 mg, v kombinácii s prednizolón
- Intravenózne podanie vysokých dávok melfolánu (100 – 200 mg/m² povrchu tela počas dvoch dní) s následnou transplantáciou autológnych kmeňových buniek
- IV dexametazón 40 mg počas štyroch dní každé tri týždne počas ôsmich cyklov
- Intravenózne podanie dexametazónu v dávke 40 mg v prvý-štvrtý, 9-12 a 17-20 deň 35-dňového cyklu, tri až šesť cyklov, po ktorom nasleduje použitie a-interferónu v dávke 3- 6 miliónov jednotiek trikrát denne týždenne
- Schéma vinkristín-doxoribucín-dexametazón (VAD).
